| DERMATOLOGÍA
(Segunda Parte)
Enfermedades Ampollares: Bases de Diagnóstico
y Tratamiento
Dr. José G.
Catacora (*)
Introducción
Se entiende como "Enfermedades Ampollares" a aquellas en las
que predominan las lesiones de contenido líquido claro, sean ampollas
o vesículas, que al romperse o secarse dejan erosiones o costras.
En su mayoría son autoinmunes y por tanto adquiridas, y cuando
son generalizadas requieren atención inmediata por lo que con
frecuencia es el médico general o médico internista el
que atiende primero a estos pacientes. Aunque no son frecuentes, sus
posibles complicaciones e incluso riesgo de muerte motiva a tener los
conocimientos necesarios para su reconocimiento y atención adecuada
(1,2). Un caso típico de enfermedad ampollar generalizada se observa
en la foto 1.
| Foto
1 |
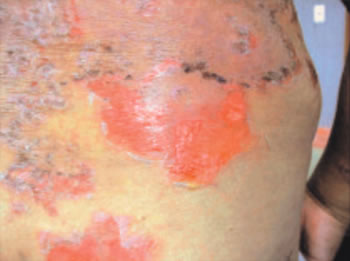 |
| Pénfigo vulgar generalizado. |
|
Existen enfermedades ampollares que se presentan al nacer o en la infancia
y no se consideran en este capítulo. De otro lado, cabe mencionar
que en muchas enfermedades que no son primariamente ampollares como la
vasculitis o el liquen plano, si la severidad del trastorno inflamatorio
es suficiente o existen condiciones locales predisponentes como trauma
cutáneo, pueden desarrollarse ampollas o vesículas en forma
secundaria, lo que podría dar lugar a confusión (3). No
debe olvidarse que varios medicamentos pueden causar reacciones adversas
con formación de ampollas (4).
Definiciones
Una vesícula es una lesión elevada, bien delimitada, de
forma oval o circular, de superficie translúcida que permite ver
un contenido líquido transparente o turbio pero no purulento,
y miden menos de 5 mm en niños y menos de 10 mm en adultos. Si
el contenido es purulento se le llama pústula.
La ampolla (o "bula"): Lesión de características
similares a la vesícula pero de dimensiones mayores.
El contenido puede ser hemático y aunque puede sugerir una formación
profunda de la ampolla, no tiene importancia desde el punto de vista
diagnóstico diferencial.
Con el tiempo las lesiones pueden evolucionar y cambiar su contenido
a pustular, o romperse dejando erosiones.
Una erosión es una lesión húmeda, o rezumante, "denudada",
generalmente sensible, resultado de rompimiento y desprendimiento del
techo de una ampolla o vesícula de las que conserva su forma.
Usualmente las erosiones alternan con las lesiones ampollares, pero pueden
ser las únicas lesiones presentes, en cuyo caso no debe dejar
de plantearse la posibilidad de una enfermedad ampollar primaria. En
el caso de cuadros de evolución prolongada, las lesiones eventualmente
remiten dejando con frecuencia máculas o manchas hiper o hipopigmentadas,
que no deben distraernos de las lesiones más importantes (5).
Aún si en el momento de evaluar por primera vez al paciente no
se pueda hacer un diagnóstico, el adecuado registro semiológico
de las lesiones cutáneas permite interpretar mejor la evolución
del paciente. Además, un correcto reconocimiento lesional facilita
la elección de la o las lesiones, que se escogerán para
la toma de muestras para estudios de diagnóstico como exámenes
directos o biopsias. Se debe prestar especial atención a la presencia
o no de pápulas o placas, que son lesiones elevadas, palpables,
sin contenido líquido y de diverso color, lo que puede ser de
utilidad en el diagnóstico como se verá posteriormente.
En esta revisión se hará referencia básicamente
a las enfermedades ampollares adquiridas.
Clasificación de las Enfermedades
Ampollares Adquiridas
Según el nivel de formación de la ampolla o vesícula
en la piel, las enfermedades ampollares adquiridas pueden dividirse en
intraepidérmicas y subepidérmicas lo que se puede ver con
mayor detalle en la tabla 1. Los pacientes con enfermedades ampollares
subepidérmicas presentan ampollas intactas y tensas con mayor
frecuencia que los pacientes con pénfigo, ya que en éstos últimos
las ampollas son más superficiales y tienden a romperse con facilidad
(6).
| TABLA 1 |
| CLASIFICACIÓN DE LAS
ENFERMEDADES AMPOLLARES |
|
| Intraepidérmicas |
Subepidérmicas
|
|
PÉNFIGOS:
- Pénfigo vulgar
- Pénfigo foliáceo
- Pénfigo eritematoso
- Pénfigo vegetante
- Pénfigo por IgA
- Pénfigo paraneoplásico
- Pénfigo inducido por drogas
|
- Penfigoide
Ampollar
- Penfigoide de las mucosas
o cicatricial
- Penfigoide gestacional
(Herpes gestationis)
- Dermatosis Ampollar por IgA
lineal (niños y adultos)
- Dermatitis Herpetiforme o
Enfermedad de Dühring-Brocq
- Epidermilosis Ampollar Adquirida
- Lupus Eritematoso Ampollar
|
|
En general se puede decir que la importancia de la diferenciación
entre una enfermedad ampollar intraepidérmica y una subepidérmica
radica en el diferente pronóstico y enfoque terapéutico
(2). Los pénfigos y en especial el pénfigo vulgar tienen
un pronóstico más reservado y requieren tratamiento más
enérgico que la mayoría de enfermedades ampollares subepidérmicas,
con la posible excepción del lupus eritematoso ampollar que casi
siempre indica enfermedad sistémica severa, o el penfigoide cicatricial
que puede ocasionar secuelas invalidantes aún con manejo adecuado
(7).
Además, con mayor frecuencia las enfermedades ampollares subepidérmicas
se acompañan de lesiones papulares o en placa eritematosas,
que constituyen un mejor lugar para la toma de muestra para biopsia de
piel, en especial para estudio por inmunofluorescencia que es uno de
los auxiliares más importantes en éstas enfermedades.
Diagnóstico Clínico
El clínico ante un paciente con enfermedad ampollar enfrenta
un reto difícil, y la búsqueda dirigida durante la anamnesis
y examen físico de datos clave permite generalmente una aproximación
diagnóstica adecuada (8,9). Así tenemos por ejemplo:
Edad de presentación: si es de nacimiento o en la infancia sugiere
epidermolisis ampollar congénita. En ancianos el penfigoide ampollar
es lo más frecuente (10).
Ingesta de drogas: el diagnóstico de erupción ampollar
medicamentosa generalmente es por descarte. Algunos individuos, genéticamente
predispuestos, pueden desarrollar cuadros típicos de pénfigo
o penfigoide al ingerir algunas drogas como antihipertensivos, bloqueadores
de canales de calcio, diuréticos o penicilamina, entre los más
frecuentes (4).
Síntomas: muchas enfermedades cursan con prurito, pero es más
severo en las mediadas por IgA, notablemente la Dermatitis herpetiforme.
En ésta última, puede incluso haber sensación de
dolor tipo hincadas muy finas. Es típica su desaparición
casi inmediata al administrar sulfonas tipo dapsona.
Signo de Nikolsky: Se basa en la pérdida de la cohesión
entre los queratinocitos, y es característico de los pénfigos
en especial del pénfigo foliáceo, pero puede verse también
en quemados y en pacientes con Necrolisis Epidérmica Tóxica
o síndrome de Lyell. Este signo se obtiene al aplicar presión
lateral sobre piel de aspecto normal a un centímetro de distancia
de una ampolla o erosión, o también jalando el techo roto
de una erosión, ésta se extiende fácilmente hacia
la piel sana(9).
Signo de Asboe-Hansen: también característico de los pénfigos,
se obtiene aplicando presión sobre una ampolla intacta observando
cómo la ampolla crece y se extiende hacia piel adyacente de aspecto
completamente sano.
Presencia de umbilicación: esta es una pequeña depresión
central en las vesículas o ampollas pequeñas y es muy sugestiva
de etiología viral, en especial por el virus herpes simple o varicela-zoster.
Aparición de placas eritematosas o lesiones tipo habón o roncha: Pueden ser las lesiones iniciales de lesiones subepidérmicas,
en especial penfigoide ampollar.
Presencia de lesiones de milio: Son lesiones papulares blanquecinas
redondeadas y superficiales de aproximadamente 1 mm de diámetro
que si se ubican en zonas de ampollas cicatrizadas sugiere una enfermedad
ampollar subepidérmica. Aunque clásicamente descritas en
las epidermolisis ampollares congénitas y adquiridas, no son
exclusivas de ellas (11).
Presencia de lesiones en diana o tiro al blanco, es característica
del eritema multiforme generalmente asociado al virus herpes simple y
debe buscarse especialmente en palmas o plantas; puede verse también
en el síndrome de Stevens-Johnson.
Disposición o agrupación de vesículas en racimos sobre una piel eritematosa: es típica de la infección herpética,
pero puede verse en dermatitis herpetiforme, dermatosis ampollar por
IgA lineal de niños y adultos, y también en el llamado
pénfigo herpetiforme.
Disposición o agrupación en forma anular o arciforme,
sugiere participación de IgA como en dermatitis herpetiforme,
dermatosis ampollar por IgA lineal de niños y adultos, pero puede
verse también en otras enfermedades ampollares subepidérmicas
como penfigoide ampollar.
Compromiso preferente o importante de pliegues cutáneos como
cuello, axilas e ingles, sugiere enfermedad ampollar subepidérmica,
tipo penfigoide ampollar, enfermedad por IgA lineal del adulto, e incluso
lupus eritematoso vesículo ampollar, además del llamado
pénfigo familiar benigno o enfermedad Hailey-Hailey que es un
enfermedad genéticamente determinada no autoinmune.
Presencia de lesiones en mucosas: no son frecuentes en pénfigo
foliáceo ni en penfigoide ampollar, sí en pénfigo
vulgar, pénfigo paraneoplásico y en penfigoide cicatricial,
además de eritema multiforme (12, 13).
| CUADRO 1 |
PATRONES HISTOLÓGICOS E INMUNOPATOLÓGICOS
DE LAS
ENFERMEDADES AMPOLLARES INTRAEPIDÉRMICAS |
|
| Enfermedad |
Histología
|
antígeno |
IFD |
IFI
|
|
Pénfigo vulgar
|
Acantolisis
suprabasal
|
desmogleína
3
|
SIC: IgG,
C3 en
capas profundas
|
% variable de(+) |
| Pénfigo foliáceo |
Acantolisis subcorneal
e intracorneal |
desmogleína 1 |
SIC: IgG, C3 en
capas superficiales |
% variable de(+) |
| Pénfigo eritematoso |
Acantolisis en capa
granulosa |
desmogleína 1 |
SIC: IgG, C3; además UDE: IgG, C3 |
% variable de(+)
20% AAN (+) |
| Pénfigo IgA |
Pústula neutrofílica
subcorneal o suprabasal
acantolisis escasa |
Desmocolina 1
desmogleína 3? |
SIC: IgA |
IgA (+) |
| Pénfigo paraneoplásico |
Acantolisis suprabasal;
queratinocitos necróticos |
Antígenos 250,
230, 210, 190 y 170 kD. |
SIC: IgG
UDE: C3 granular |
IgG(+) anti SIC
epitelios simples |
Pénfigo Famlilar benigno
(Enf.Hailey-Hailey)* |
Acantolisis suprabasal
e intraepidérmica |
negativo |
negativo |
negativo |
|
SIC:sustancia intercelular; UDE: unión
dermo epidérmica; AAN: anticuerpos antinucleares
*no es una enfermedad autoinmune sino genética |
| CUADRO 2 |
PATRONES HISTOLÓGICOS E INMUNOPATOLÓGICOS
DE LAS
ENFERMEDADES AMPOLLARES SUBPIDERMICAS |
|
| Enfermedad |
Histología
|
antígeno |
IFD |
IFI
|
IFI
piel despegada |
IME |
|
Pénfigoide Ampollar
|
Ampolla subepidérmica, eosinófilos
|
BP230, BP 180
|
UDE: IgG, C3 lineal
|
Hasta 75%(+) IgG en UDE |
Unión en porción epidérmica |
inmunorreactantes en lámina lúcida |
| Penfigoide Gestacional |
Ampolla subepidérmica, eosinófilos,
espongiosis
|
BP 180 |
UDE: C3 100%
IgG 40% |
10% IgG en UDE
100% factor HG, fijador complem. |
Unión en porción epidérmica |
C3 e IgG en lámina lúcida |
| Penfigoide de las Membranas Mucosas |
Ampolla subepidérmica |
BP 230, BP 180, Laminina 5 |
UDE: IgG lineal 80% IgA 40% |
10% IgG en UDE
IgA: forma ocular |
variable, dérmica y/o epidérmica |
variable, lámina lúcida
o densa, hemidesmosomas |
Epidermolisis ampollosa Adquirida
|
Usualmente escasa inflamación
dérmica |
Antígenos colágeno VII |
UDE: IgG, IgM, C3 patr?2n lineal |
25% IgG en UDE |
Unión en porción epidérmica |
inmunorreactantes en sublámina
lúcida |
| Dermatitis herpetiforme |
Microabscesos papilares neutrofílicos |
|
IgA granular 100% en papilas C3 40% |
20% IgG en UDE |
No aplicable |
|
| Enf. Ampollar IgA lineal |
Infiltrado neutrofilico |
Antígeno 97kD, colágeno VII, BP 180,
BP 230 |
UDE: IgA 80% |
Niños: 75% IgA UDE
adultos: 20% IgA UDE |
Unión en porción epidérmica |
inmunorreactantes en porción epidérmica,
pocos en porción dérmica |
|
| BP: antígeno de penfigoide buloso (o
ampollar); unión dermo epidérmica. |
Confirmación del diagnóstico
Son muchas las pruebas empleadas para confirmar el diagnóstico
pero la más importante es el estudio histológico e inmuohistoquímico
de piel (9).
Las biopsias de piel deben ser en lo posible múltiples y de diversos
sitios que incluyen: borde de ampolla (o vesícula íntegra
si su tamaño y el instrumental y experiencia lo permiten) para
observar el nivel de formación de la lesión o sea intra
o subepidérmica. El borde de ampolla o vesícula puede no
ser el mejor lugar para estudio de inmunofluorescencia directa (IFD).
Para IFD es mejor escoger una lesión papular o urticariana, o
en caso de no haber éstas, entonces la piel no lesional adyacente
a las lesiones ampollares puede servir (14, 15).
Como éstos estudios pueden tomar algunos días, un forma
rápida de confirmar inicialmente un pénfigo es mediante
el test de Tzanck en que se observaría las células epidérmicas
acantolíticas de forma redondeada, permitiendo de paso descartar
una infección herpética que causa cambios citopáticos
característicos (células gigantes multinucleadas).
En los cuadros 1 y 2 se observan los patrones histológicos e
inmunohistoquímicos diferenciales de las enfermedades ampollares.
Estos estudios son metodológicamente laboriosos y realizados
en centros de referencia.
Manejo inicial del paciente
con Enfermedad Ampollar
El enfoque inicial deberá considerar dos puntos esenciales, el
estado general del paciente, incluyendo posible sepsis, y la extensión
de las lesiones ampollares, en especial si se han roto dejando superficies
abiertas. Una enfermedad localizada generalmente tiene poco riesgo, pero
en caso de ser generalizada el riesgo aumenta sensiblemente al aumentarse
las probabilidades de sepsis originada en piel, así como los disturbios
metabólicos serios por pérdida de agua y electrolitos,
con alteraciones del equilibrio ácido básico, y si hay
inmovilidad prolongada riesgo de trombosis venosa profunda y pulmonar.
Por ejemplo la mortalidad del pénfigo vulgar aún tratado
adecuadamente puede oscilar entre el 5% y 10%.
El plan de trabajo será orientado a confirmar el diagnóstico,
alejar los diagnósticos diferenciales, evaluar complicaciones
posibles y enfermedades preexistentes, y preparar al paciente para eventual
terapia inmunosupresora. Los casos muy severos deben ser manejados por
un equipo multidisciplinario capacitado con experiencia, que incluya
enfermería, nutrición, sicología, asistencia social,
además de las especialidades médicas necesarias usualmente
bajo la dirección del dermatólogo.
En la tabla 2 se presenta un plan de manejo sugerido si corresponde
recibir a un paciente con enfermedad ampollar generalizada.
| CUADRO 2 |
PLAN DE TRABAJO Y MEDIDAS INICIALES EN
CASOS DE ENFERMEDAD AMPOLLAR GENERALIZADA |
|
- Registro de signos
vitales.
- Colocación de catéter venoso
a través
de piel no lesional, evitando esparadrapo u otro material
adhesivo.
- Evaluación de la extensión de
la enfermedad: regla de nueves.
- Registro detallado de
medicación previa y suspensión
de lo sospechoso.
- Estudios microbiológicos: hemocultivos,
urocultivos, cultivo de piel para gérmenes comunes.
- Toma de biopsias de piel y test
de Tzanck si corresponde.
- Estudio hematológico
y bioquímico funcional
hepático y renal, electrolitos séricos, perfil
de coagulación.
- Estudios radiológicos
básicos.
- Descarte de otros posibles focos infecciosos,
por ejemplo parásitos intestinales.
- Consultas:
dermatología, oftalmología,
soporte nutricional de estar disponible, entre otras.
- Inicio
de antibioterapia antiestafilocócica si
presenta fiebre u otro signo de sepsis: taquicardia, aumento
de la frecuencia respiratoria, alteración del sensorio,
reducción de diuresis.
- Medidas generales:
- Ubicación en ambiente aislado y con ropa estéril.
- Desinfección de manos y empleo de guantes y medidas
de protección durante examen.
- Limpieza de la piel o baño en lo posible 2 veces al día, y
aplicación
de
antibióticos locales tipo mupirocina o ácido fusídico,
con la precaución de
no administrar sulfadiazina si se
sospecha S. de Lyell que con cierta frecuencia es causado por sulfas.
- Alimentación oral salvo extrema incapacidad por compromiso bucal o
esofágico,
con aporte de acuerdo a estado general considerando hipercatabolia.
- Monitorización de signos vitales y diuresis.
- Anticoagulación profiláctica.
|
|
Tratamiento
El tratamiento en general dependerá del diagnóstico específico
y del estado general del paciente considerando su respuesta previa a
otros tratamientos. Los casos recalcitrantes por ejemplo requieren un
enfoque diferente.
La piedra angular del tratamiento de las enfermedades ampollares autoinmunes
son los corticosteroides y los inmunosupresores además de otras
drogas. Su manejo y complicaciones van más allá del alcance
de esta revisión. Un breve resumen de las opciones terapéuticas
en algunas de ellas se muestra en el cuadro 3.
| CUADRO 3 |
PATRONES HISTOLÓGICOS E INMUNOPATOLÓGICOS
DE LAS
ENFERMEDADES AMPOLLARES INTRAEPIDÉRMICAS |
|
| Enfermedad |
Elección |
Secundaria |
Otras Opciones |
|
Pénfigo Generalizado
|
Corticosteroides prednisona o metilprednisona:
1 a 2.5 mg/k peso/día
|
Asatioprina
Ciclofosfamida
metotrexato
micofenolato
sales de oro
ciclosporina
|
Terapia pulsar EV, VO con corticosteroides
o ciclofosfamida
Plasmaferesis
ganaglobulinas intravenosa fotoféresis extracorpórea
terapia inmunoablativa
|
| Pénfigo localizado |
Corticosteroides prednisona similar dosis
bajas o alternas |
Corticoides
tópicos |
Sólo inmunosupresores como azatioprina |
| Pénfigo ampollar generalizado |
prednisonia o metilprednisona: 0.5 a 1
mg/k peso/día |
igual a penfigo Clobetasol propionaso tópico |
similar a pénfigo |
| Pénfigoide cicatricial severo |
prednisona 1mg/k/d |
Azatioprina otros inmunolosupresores |
Terapia pulsar EV, VP con corticosteroides
o ciclofosfamida |
|
Conclusiones
Las enfermedades ampollares son un grupo heterogéneo de dermatosis
en las que se han realizado grandes avances en el conocimiento de su
patogénesis y tratamiento.
A pesar de no ser un problema frecuente,
los pacientes con enfermedades ampollares en especial si son generalizadas
se encuentran en riesgo de
complicaciones potencialmente riesgosas, y un adecuado manejo desde el
inicio puede ayudar a mejorar su pronóstico.
Bibliografía
- Crosby, DL; Díaz, LA. Introduction to bullous dermatoses.
Dermatol Clin 1993;11(3):373-78.
- Yeh, SW; Ahmed, B; Sami, N; Ahmed,
RA. Blistering disorders: diagnosis and treatment. Dermatol Ther
2003;16(3):214-23.
- Shahab, RK; Loo, DS. Bullous scabies. J Am Acad Dermatol
2003;49(2):346-50.
- Differential diagnosis of severe cutaneous drug
eruptions. Am J Clin Dermatopathol 2003;4(8):561-72.
- Goldsmith, L;
Lazarus, G; Tharp, M. Adult and Pediatric Dermatology. 2nd. Ed. Philadelphia;
F.A. Davis Company, 1997.
- Devries, DT; Warren, SJ. Recent advances
in intraepidermal blistering diseases. Adv Dermatol 2003;18:203-45.
- Stoopler,
ET; DeRossi, SS; Sollecitto, TP. Mucous membrane pemphigoid: Update
for the general practitioner. N Y State
Dent J 2003;69(8):28-31.
- Maharshak, N; Brenner, S. Gender
differences in vesiculobullous autoimmune skin diseases. Skinmed
2002;1(1):25-30.
- Mutasim, DF; Pelc, NJ; Supapannachart, N. Established
methods in the investigation of bullous diseases. Dermatol Clin
1993;11(3):399-418.
- Mutasim, DF. Autoimmune bullous dermatoses
in the elderly: diagnosis and management. Drugs Aging 2003;20(9):663-81.
- Trent,
JT; Kirsner, RS. Epidermolysis bullosa identification and treatment.
Adv Skin Wound Care 2003;16(6):284-90.
- Ayangco, L; Rogers, RS. Oral
manifestations of eythema multiforme. Dermatol Clin 2003;21(1):195-205.
- Robinson,
NA; Wray, D. Desquamative gingivitis: a sign of mucocutaneous disorders-a
review. Aust
Dent J 2003;48(4):206-11.
- Korman, N. Bullous pemphigoid. J Am Acad
Dermatol 1987;16:907.
- Korman, N; Pemphigus, J. Am Acad Dermatol 1988;18:1219.
(*)
Jefe del Servicio de Dermatología del Hospital Nacional Guillermo
Almenara Irigoyen (HNGAI) - EsSalud.
Profesor por Asignatura de Dermatología de la Universidad Peruana
Cayetano Heredia (UPCH). |