Síndromes Anémicos (primera parte)
Anemias Carenciales
Dr. Oscar Ruiz Franco (*)
Anemía Ferropénica
Es la anemia más frecuente en el mundo y se presenta sobretodo en los países pobres. Para que se evidencie la anemia ferropénica es necesario que se cumplan las siguientes etapas: a) ferropenia pre-latente: disminución hierro medular; b) ferropenia latente: disminución de ferritina y saturación de transferrina y, c) eritropoyesis ferropénica: anemia, microcitosis e hipocromia (1, 2, 3).
Patogenia:
Eritropoyesis Ferropénica
El Islote Eritroblástico es la unidad funcional fundamental para la entrada del hierro de los macrófagos medulares a los eritroblastos. Está conformada por un macrófago rodeado de eritroblastos. Mediante la rofeocitosis, el macrófago cede el hierro a los eritroblastos ya que por sí solos, estos no pueden incorporar directamente el hierro sérico (2).
Los requerimientos diarios de hierro de un organismo sano para cubrir las necesidades de la eritropoyesis varían durante el crecimiento y por efectos de la menstruación, embarazo y lactancia. La formación de 1 mL de eritrocitos precisa de 1 mg de hierro, por lo tanto la eritropoyesis consume cada día entre 20 y 25 mg. El hierro corporal total varía entre 3 y 4 gr, de los cuales, el 80% sirve para la síntesis de hemoglobina y el 20% se encuentra como depósito (en la forma de ferritina y hemosiderina). Las consecuencias de la eritropoyesis ferropénica son el desarrollo de microcitosis e hipocromía producto de la disminución de la síntesis de hemoglobina por la falta de hierro. Además, disminuye la actividad de proteínas que contienen hierro: citocromos, mieloperoxidasa (2).
Las causas más frecuentes de ferropenia son: ingesta inadecuada, malabsorción, pérdida crónica, desviación del hierro de la madre al feto/ lactante y hemólisis intravascular (2).
Cuadro Clínico
• Alteraciones debidas a ferropenia: disfunción neurológica con disminución del rendimiento intelectual, irritabilidad, cefalea y pica. Disminución de la secreción de ácido gástrico. Se puede presentar, además, atrofia de la mucosa oral, vía gastrointestinal, piel y faneras (3, 5).
• Síntomas Específicos de ferropenia: a) en boca: glositis, estomatitis, queilitis comisural; b) en uñas: platoniquia, coiloniquia; c) en cabellos: alopecia, sequedad; d) pica (ingesta inusual de sustancias, por ejemplo, papel, tierra, hielo, etc); e) en estómago: dispepsia (3, 5).
Exámenes de Laboratorio
Hemograma
• Eritrocitos: En la serie roja se evidencia incremento del índice de anisocitosis (hematíes de diferente tamaño), Figura 1: Coloración de Pearls para valoración de hemosiderina. Arriba, depósitos normales de hemosiderina que se aprecian de color verde azulado sobre un fondo amarillo. Abajo, ausencia en los depósitos de hemosiderina, la espícula aparece vacía.microcitosis (VCM <80 fL), hipocromía (HCM <27 pg). Además, en el frotis de sangre periférica se pueden encontrar células en habano o lápiz y dianocitos (3).
• Leucocitos: Puede haber tanto leucopenia como leucocitosis (3).
• Plaquetas: En la serie plaquetaria lo más frecuente es el hallazgo del trombocitosis (35% en niños y hasta 75% en adultos) mientras que la trombocitopenia es mas frecuente en niños (28%). Todas estas alteraciones desaparecen con el tratamiento de la anemia (4, 5).
| FIGURA 1 |
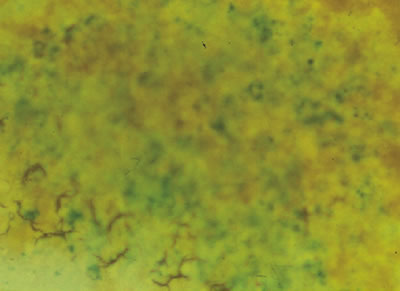 |
| Figura 1: Coloración de Pearls para valoración de hemosiderina. Arriba, depósitos normales de hemosiderina que se aprecian de color verde azulado sobre un fondo amarillo. Abajo, ausencia en los depósitos de hemosiderina, la espícula aparece vacía. |
Médula ósea
Los depósitos de hierro (hemosiderina) pueden estar ausentes o disminuidos (Figura 1). Su evaluación es el parámetro más confiable para el diagnóstico de anemia ferropénica. En relación a la celularidad hematopoyética, ésta se halla, generalmente, incrementada con predominio de la serie eritroide(2).
Metabolismo del Hierro
En la actualidad se constituye como el método diagnóstico más usado. Evalúa el hierro sérico, que en este caso se halla disminuido; la capacidad total de unión de la transferrina (CTST) se encuentra incrementada; y, la ferritina sérica, disminuida. El hallazgo de una ferritina menor de 10 ng/ mL nos induce a pensar en anemia ferropénica (2).
Tratamiento
El tratamiento abarca tres aspectos: etiológico, sintomático y preventivo (2, 5, 6).
• Tratamiento etiológico: en lo posible se debe eliminar la causa que produce la anemia ferropénica. Por ejemplo, ante una hipermenorrea, ésta puede reducirse con la administración de anovulatorios. Las causas digestivas ya sean benignas malignas, generalmente exigen un tratamiento quirúrgico.
• Tratamiento sintomático: Tiene dos finalidades, incrementar la concentración de hemoglobina y restaurar los depósitos de hierro.
• Tratamiento preventivo: Tiene como finalidad evitar la aparición de ferropenia. Debido a que la prevalencia es prácticamente igual en todas las poblaciones del mundo y dado que existen grupos de alto riesgo (niños y mujeres jóvenes), éste compete a las autoridades sanitarias de todos los países.
El tratamiento consiste en el aporte suplementario de hierro a las poblaciones de alto riesgo. Países desarrollados como Estados Unidos y Suecia, enriquecen la harina con 29 a 36 mg de hierro por kilo y con ello han conseguido disminuir el porcentaje de individuos afectados por ferropenia. La OMS recomienda la realización de programas de fortificación global o dirigido a las poblaciones en alto riesgo (2).
Vía Oral
La administración de hierro debe hacerse preferentemente como sales ferrosas debido a su mejor absorción intestinal y bajo costo. Una dosis diaria debe aportar entre 100 y 200 mg de hierro elemental, la cual puede prescribirse a razón de 2 a 3 tabletas a lo largo del día, siempre antes de la ingesta de comidas. En el estado ferropénico, la absorción intestinal de hierro (normal: 2,5 a 10%) puede incrementarse hasta 30%, por lo tanto, la dosis citada, permite el ingreso de 20 a 40 mg de hierro diariamente (2, 5).
La respuesta favorable se manifiesta con un aumento de reticulocitos que se inicia alrededor del cuarto día del tratamiento y persiste por doce días. Simultáneamente, hay un incremento progresivo de hemoglobina (0,15 gr/L/día) hasta alcanzar los valores normales a las 4 ó 10 semanas de tratamiento. Luego de restaurar el valor normal de hemoglobina, debe continuarse con la administración de dosis de alrededor de 100 mg por día durante 2 a 3 meses para restituir los depósitos en la médula ósea (2, 5).
Vía Parenteral
Se recurre a ella en circunstancias en las que no se pueda utilizar la vía oral: existencia de factores que impidan la absorción como cirugía gastro intestinal, hemorragia digestiva activa o intolerancia a los preparados de hierro oral. La dosis total de hierro parenteral a administrar durante todo el tratamiento se calcula mediante la siguiente fórmula (2, 4, 8):
Fe (mg) = (Hbi - HbR) x Peso (Kg) x 2,2 + 1000
HbI: Hemoglobina ideal, por lo común ésta es 15.
HbR: Hemoglobina del paciente al momento del diagnóstico.
1000: Representa los depósitos de hierro a saturar.
Anemia Megaloblástica
Este tipo de anemia es causada por la disminución en la síntesis de ADN, debido a deficiencia de folato y/o vitamina B12. Cuando se analiza la posible influencia de la dieta en la aparición de una deficiencia vitamínica es importante tener en cuenta que para la cobalamina, las reservas pueden subsistir entre 3 y 5 años sin aporte vitamínico, mientras que las de ácido fólico se agotan en 3 a 4 meses (1-3).
Las anemias megaloblásticas se caracterizan por detención en la maduración y por la presencia de células grandes con núcleo inmaduro, alteración que se halla presente en las tres series celulares de la médula ósea (2-4).
Fisiopatología
Como se mencionó anteriormente, la anemia megaloblástica es expresión del transtorno madurativo de los precursores de la eritropoyesis debido a la falta de cobalamina y ácido fólico (vitaminas esenciales para la síntesis del ADN) (2).
• Hematopoyesis normal: Las células con capacidad de autoduplicarse se encuentran la mayor parte del tiempo en reposo (fase del ciclo celular G0). En la fase de mitosis (S), las células duplican su contenido de ADN y vuelven a la fase de reposo (2, 9).
• Hematopoyesis megaloblástica: La fase S está prolongada, por ende hay más células intentando duplicar su contenido de ADN que en fase de reposo; esto se traduce en la aparición de células grandes con cromatina reticulada. La falta de folato y/o vitamina B12 altera la formación del ADN, pues se produce una menor conversión de uridina en timidina. Este transtorno del ADN se acompaña de síntesis normal de hemoglobina, por lo tanto no hay hipocromia (2, 9).
La anemia megaloblástica carencial puede acompañarse de alteraciones de células no hematopoyéticas con elevado recambio celular (piel, mucosas, epitelio gastro intestinal) con aparición de lesiones tróficas o inflamatorias de la piel y mucosas (2, 9).
• Causas de anemia megaloblástica: Las causas más importantes son: ingesta inadecuada, mala absorción, incremento de los requerimientos, uso de fármacos (anti-neoplásicos, quimioterápicos). En nuestro país, la etiología es multifactorial debido a que la mayoría de la población no satisface los requerimientos mínimos diarios de vitaminas y oligoelementos y, además, porque el acceso a los servicios de salud básica es muy limitado. Esto se torna más dramático en las provincias más alejadas como por ejemplo sucede en la zona del trapecio andino (9).
Cuadro Clínico
Como síntomas específicos de anemia megaloblástica podemos citar entre los más importantes: piel seca y amarillenta, ictericia leve, glositis atrófica caracterizada por pérdida de papilas gustativas y aumento de la sensibilidad dolorosa, ulceraciones, alteraciones en la percepción del gusto. Además es usual encontrar cuadros de diarrea crónica y dispepsia (2,3,4,8,9).
Mención aparte merecen las manifestaciones neurológicas, las cuales se presentan sólo en casos de falta de vitamina B12. Así, los pacientes suelen presentar parestesias, disminución de la sensibilidad superficial, deambulación inestable, disminución de la sensibilidad profunda, incoordinación, signo del Romberg positivo, pérdida de la fuerza muscular, hiperreflexia, espasticidad
clonus y signo de Babinsky bilateral (2, 4, 9).
Requerimientos
Folato : 50 a 100 µg / día
Vitamina B12 : 2 a 5 µg / día
Exámenes de Laboratorio
• Hemograma: En la serie roja se evidencia incremento del índice de anisocitosis (hematíes de diferente tamaño), macrocitosis (VCM > 100 fL). En el frotis de sangre periférica se pueden encontrar ovalocitos, dacriocitos, cuerpos de inclusión (Howell Jolly, anillos de Cabot). En el caso de los leucocitos, sólo alteran su recuento en condiciones carenciales severas, en cuyo caso puede aparecer leucopenia con granulocitopenia intensa. Otros hallazgos son la presencia de neutrófilos grandes e hipersegmentados (> 5%) (9, 10).
Finalmente, el recuento plaquetario generalmente no suele verse alterado pero puede cursar con trombocitopenia severa. Es característica la presencia de macroplaquetas. Cabe mencionar que las anemias megaloblásticas son una causa relativamente frecuente de pancitopenia, por lo que se debe hacer el diagnóstico diferencial con aplasia medular (9 10).
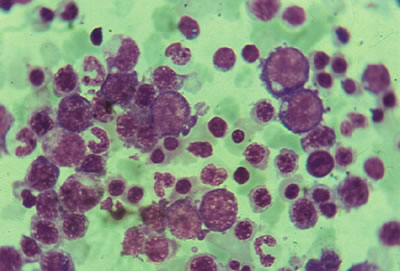
• Médula ósea: Los depósitos de hierro generalmente están incrementados pero también pueden encontrase dentro de límites normales. La celularidad hematopoyética siempre se encuentra incrementada mostrando una gran detención el proceso de maduración de las tres series, siendo ésta mas evidente en la serie eritroide. La célula más característica la constituye el megaloblasto, el cual se caracteriza por tener un tamaño superior al normal y una cromatina finamente reticulada, propia del retraso madurativo nuclear. Así, mismo aparece asincronía en la relación entre el núcleo y el citoplasma a favor del núcleo (1, 2, 8).
• Bioquímica: En el suero se pueden determinar directamente los niveles de folato y vitamina B12, los cuales deben encontrarse disminuidos. En el caso de anemia megaloblástica por carencia de folato, es recomendable la determinación del folato eritrocitario, pues es mas específico al no encontrarse afectado por la influencia de la dieta (2, 8, 9).
Figura 2.- Médula ósea con maduración megaloblástica. Nótese el marcado predominio de la serie eritroide así como la presencia de megaloblastos (células grandes, mono nucleadas con el borde de la membrana citoplasmática muy irregular).
| Figura 2.- Médula ósea con maduración megaloblástica. Nótese el marcado predominio de la serie eritroide así como la presencia de megaloblastos (células grandes, mono nucleadas con el borde de la membrana citoplasmática muy irregular). |
Anemia Perniciosa
La expresión clínica más precoz está constituida por tres síntomas: pérdida progresiva de la fuerza muscular, especialmente en extremidades inferiores; lengua inflamada y dolorosa; y, signos neurológicos consistentes en parestesias y Babinsky positivo. El síndrome neurológico que acompaña a la anemia perniciosa se le conoce como la degeneración combinada de los cordones laterales y posteriores de la médula espinal. La exploración física muestra palidez de piel y mucosas, alteraciones de la pigmentación cutánea (vitíligo o melanodermia). Esta enfermedad es de naturaleza autoinmune, por ello se puede asociar a hipo o hipertiroidismo, insuficiencia córtico suprarenal, diabetes o hipoparatiroidismo. La respuesta al tratamiento es similar al de las otras formas de anemia megaloblástica, es decir, mediante la evaluación de la reticulocitosis(2, 9).
Pruebas Diagnósticas
Son fundamentalmente tres: prueba de absorción de la cobalamina, determinación de anticuerpos anti-factor intrínseco y análisis de la mucosa gástrica (2, 9).
• Prueba de Schilling: Consiste en cuantificar la excreción urinaria de cianocobalmina marcada con cobalto el cual es ingerido por vía oral (1 µg con un radioactividad de 0,5 mCu), previa saturación del transportador mediante inyección intramuscular de 1000 µg de cobalamina no marcada. La orina es colectada durante 24 horas y en condiciones normales, debe contener más del 5% de la dosis de cobalamina marcada; los pacientes con mala absorción generalmente eliminan menos del 2%. En el caso de anemia perniciosa, la prueba de Schilling será positiva cuando la eliminación de cianocoblamina marcada sea mayor del 5% (9-10).
• Anticuerpos Antifactor Intrínseco: Su hallazgo constituye la prueba de mayor valor en el diagnóstico de anemia perniciosa, ya que su demostración es altamente específica de gastritis atrófica. Son anticuerpos de tipo IgG altamente específicos pero poco sensibles, pues se detectan en algo más del 50% de casos de anemia perniciosa. Otros anticuerpos que se detectan con mayor frecuencia (> 80%) están dirigidos contra la mucosa gástrica (anti parietales y anti mucosa gástrica); éstos tienen el inconveniente de ser poco específicos, pues se pueden encontrar también en otras enfermedades de origen inmune (2, 4, 9).
• Examen Histológico de la mucosa gástrica: consiste en el estudio macroscópico y microscópico de la mucosa gástrica. La biopsia revelará la ausencia casi absoluta de células parietales y principales, con focos de infiltración linfocitaria. Este hallazgo es de gran valor para el diagnóstico de la enfermedad (4, 9).
Tratamiento
Esta íntimamente ligado a la causa que la produce y generalmente responde bien a la administración de la vitamina deficiente. La evaluación del tratamiento se realiza con la respuesta reticulocitaria, la cual se evidencia a partir del segundo día, alcanzando su cifra más alta en los días 5 y 6 del tratamiento y persiste hasta el día diez. Puede aparecer un nuevo incremento en los reticulocitos luego del día diez pero sin llegar a alcanzar la magnitud del primero(1, 2, 8).
Tratamiento de la deficiencia de folato
Puede ser éste de dos tipos, preventivo o curativo. El tratamiento curativo consiste en la administración oral de 50 a 100 µg por día hasta que desaparezcan las manifestaciones clínicas o hematológicas de la megaloblastosis. Si no se observa respuesta, se debe emplear la vía parenteral 5 mg/ mL.
El tratamiento preventivo es recomendado en situaciones donde existe hiperconsumo de folato (embarazo, niños prematuros, pacientes sometidos a hemodiálisis, cuadros hemolíticos de larga duración). La administración profiláctica debe ser de 0,2 a 0,4 mg/ día. En pacientes sometidos a tratamiento citostáticos es preferible emplear ácido folínico por vía parenteral a dosis de 3 a 15 mg/ día (2, 3).
Tratamiento de la Deficiencia de vitamina B12
Tiene como objetivo corregir la anemia y las alteraciones epiteliales. Reducir los transtornos neurológicos o prevenir su aparición y, normalizar los depósitos hísticos de cobalamina. La administración debe realizarse por vía parenteral, mediante dosis de 1000 µg durante una semana y dosis de mantenimiento de 1000 µg, una vez al mes.
La primera dosis restituye los depósitos y corrige la anemia mientras que la segunda dosis, se mantiene durante el tiempo que sea necesario según la naturaleza de la enfermedad que provocó la deficiencia de vitamina B12. En el caso de anemia perniciosa el tratamiento se mantiene por toda la vida con vigilancia periódica de la mucosa gástrica para detectar precozmente la aparición de un probable carcinoma (2-4).
Bibliografía
- Lee et al . "Wintrobe's Clinical Hematology". 10 ma Edición. Williams & Wilkins. Houston. 1999.
- Sans Sabrafen y col. "Hematología clínica". 4ta Edición. Editorial Hartcourt. Madrid España. 2001.
- Beuler, Ernest,et al. " Williams Hematology". 5ta Edición. Editorial Mc Graw - Hill. 1995.
- Weiner, M. et al. "Pediatric Hematology/Oncology Secrets". 2 da Edición. Editorial Hanley & Belfus. 2002.
- Wood, M. y col. "Secretos de la Hematología y Oncología". 2da Edición. Editorial Interamericana. 2000.
- De Mayer et al. "The influence of anemia in the world". WHO Reports. 1978;38:302-16.
- Cook, JD. "Clinical evaluation ofniron deficiency". Seminars in Hematology. 1978;19: 6-21.
- De Gruchy, GC. "Clinical hematology en medical practice" 4ta Edición. Editorial Blackwell. Oxford. 1978.
- Chanarin, I. "The megaloblastic anemias". 3ra edición. Editorial Blackwell. Oxford. 1990.
- Shilling,R.F. “Intrinsic factor studies II. The effect of gastric juice on the urinary excretion of radioactivity after oral administration of radioactive vitamin B12”. J Lab clin Med 1953;42.
- Crowe, SF. "Effect of folate deficiency and folate and B12 excess on memory functioning in young chicks". Pharmacological Biochemical Behavior. 1997;56:189-97.
(*) Profesor Asociado Facultad de Medicina Universidad Nacional Mayor de San Marcos (UNMSM).
Jefe del Servicio de Hematología Clínica del Hospital Nacional “Dos de Mayo”.
|