|
|
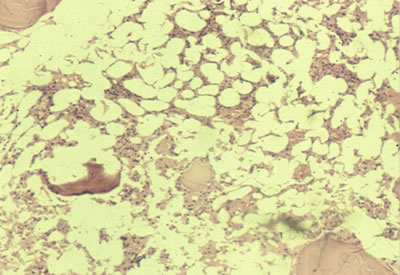
Síndromes Anémicos (primera parte) Dr. Norberto Quezada Velásquez (*) La primera descripción sobre la enfermedad corresponde a Erlich en 1888, en la que comunica un caso de anemia, leucopenia, trombocitopenia y ausencia de regeneración medular, en una paciente joven embarazada, que fallece como consecuencia de la anemia y la neutropenia severa, encontrando en la necropsia médula ósea grasa y ausencia de hematopoyesis. Este término, fue aplicado por muchos años, pero confundiéndose con otras patologías que cursan con pancitopenia. Blumer en 1905, objeta el término de anemia aplásica, porque muchos pacientes no mostraban hipocelularidad de la médula ósea, si no mas bien asociación con incremento linfoide. Dos años después Luzatto, describe pacientes con pancitopenia, con médula ósea normo o hipercelular, denominándolos como, “anemia seudoaplástica”. Rhoads y Baker, tratan de unificar los criterios anteriores en una revisión de cien casos, reuniendo las diversas manifestaciones hematológicas, e introducen el término de “anemia refractaria” y la dividen en primaria o idiopática y secundaria, pero considerando dentro de la segunda casos de tumores y leucemias. Bonford y Rhoads, en 1941, dan mayor certeza a la denominación de “anemía refractaria”, pero incluyendo dentro de ella casos de mielofibrosis (1). Posteriormente, se han publicado en 1982 Camitta (2) y Thomas 1984 (3), revisiones muy completas, que han permitido establecer los criterios diagnósticos definitivos. Sin embargo, se observó que la mayoría de pacientes, fallecían a los seis meses de establecido el diagnóstico, estos datos llevaron a definir criterios ya no diagnósticos que habían sido clarificados, sino parámetros que pudieran evaluar, la evolución, los cuales fueron definidos por Camita (4) y son lo que se usan internacionalmente hasta ahora. Entonces el término anemia aplásica, describe un síndrome clínico en el cual hay anemia, neutropenia, plaquetopenia, y la médula ósea muestra acentuada disminución de los precursores eritropoyéticos, con reemplazo por tejido graso. Desde la introducción de la Globulina antífinfocitica (GAL) y el transplante de médula ósea (TMO), se ha experimentado un cambio drástico en la supervivencia de los pacientes y además es posible obtener la curación en un determinado grupo los mismos. Posibles mecanismos patogénicos Hay una serie de mecanismos patogénicos, a los cuales se pueden atribuir la falla adquirida, de la médula ósea, se incluyen dentro de estos: lesión del Stem-cell, lesión del estroma de la médula ósea, virus capaces de producir agresión de las células hematopoyéticas, agentes tóxicos, mielopatía monoclonal, mecanismos celulares y humorales capaces de producir supresión de la hematopoyesis. Lesión del Stem Cell La recuperación de la hematopoyesis en el 50 %, de los pacientes con anemia aplásica severa (AAS), transplantados con médula ósea procedente de un gemelo homocigótico, sin acondicionamiento previo, puede estar representando en forma clara, que estos pacientes no tienen lesión del microambiente medular (MAM) ni alteración inmunológica (5). Lesión del microambiente de la médula ósea. Los ratones de cepa S1/S1, caracterizados por lesión del micro ambiente medular. (Está formado por losfibroblastos, mastocitos, adipocitos, células endoteliales, macrófagos y células reticulares) MAM. Se ha observado lo siguiente: la médula ósea, de estos ratones tienen capacidad para restaurar la hematopoyesis en ratones de otra cepa, irradiados letalmente. Sin embargo, la médula ósea de estos últimos no es capaz de normalizar la hematopoyesis de los primeros, lo que sugiere lesión del MAM (6). Las células del MAM, tienen un papel funndamental en el estímulo de la hematopoyesis y por lo tanto su lesión podría desarrollar una insuficiencia medular. Agentes tóxicos El benceno, fue la primera sustancia vinculada con la anemia aplásica, en un estudio de los trabajadores de una factoría alrededor de los años veinte. A pesar de sus conocidas propiedades como tóxico de la médula ósea, el benceno es todavía usado ampliamente como solvente y es empleado en la manufactura de químicos, drogas, colorantes y explosivos. Además es un importante químico en la manufactura de gomas y de zapatos. En la China donde el benceno es ampliamente usado, la incidencia de anemia aplásíca fue seis veces mayor que en la población general. Además otras anormalidades hematológicas han sido relacionadas con el benceno, como anemias hemolíticas, metaplasia mieloide y leucemia mielblástica. Hay al parecer también una relación entre el uso de insecticidas y anemia aplásica, se ha publicado en la literatura un buen número de casos asociados con compuestos órganofosforados. El DDT (clorofenotano) parece ser el más común. Nosotros encontramos en una casuística de noventa pacientes, que el 33-5 %, fueron considerados como anemia aplásica secundaria y de los cuales, 11 de ellos estuvieron relacionados con insecticidas (7). Entre las drogas se han descrito un gran número de ellas asociadas con anemia aplásica, pero cabe mencionar al cloranfenicol que posee un compuesto de nitrobenceno, este antibiótico de amplio espectro, fue introducido en 1948 y usado ampliamente, durante las décadas del 50 y 60, a menudo sin una indicación correcta. Dos años después de su introducción en 1950, Rich comunica el primer caso atribuido a esta droga. El riesgo de desarrollar anemia aplásica en pacientes tratados con cloranfenicol, es de cerca de 1 en 20,000 ó de 10 a 50 veces más que en la población general (8). Otra droga, que está perfectamente establecida como productora de lesión de la médula ósea, es la quinacrina, que se usó como antimalárico, durante la segunda guerra mundial,en misiones selváticas en el sudoeste asiático en los años de 1943 y 1944, encontrándose entre 7 a 28 casos por año por millón de personas, contra 1 a 2 por millón, en los que no la recibían. También han sido asociadas con la producción de anemia aplásica, los anticonvulsivantes, particularmente la carbamezepina, las sulfonamidas y sus derivados que incluyen a los diuréticos y agentes hipoglicemiantes. Las radiaciones ionizantes también están consideradas como agentes tóxicos sobre la médula ósea, como las exposiciones crónicas a bajas dosis de radiación localizadas, para el tratamiento de la espondilítis esclerosante. Los accidentes nucleares como el de Chernobyl y las explosiones atómicas de Nagasaki e Hiroshima son los ejemplos más claros de su acción nociva. Los virus Los virus, también son capaces de producir lesión en las células precursoras de hematopoyesis, asociadas con el desarrollo de anemia aplásica, este hecho ha sido comunicado por Young y Mortimer en 1984 (9). Los virus pueden tener una capacidad directa para lesionar las células germinales, pero su acción no está establecida, como es el caso de la hepatitis (no A, no B) el virus de Epstein-Barr y los parvos virus, dentro de los cuales se le asigna una mayor importancia al B19. La otra posibilidad es que pueden integrarse al genoma celular, alterando su contenido nuclear, creando una distorsión en la hematopoyesis. También es posible que estimulen una respuesta humoral anormal, ya que desde el punto de vista inmunológico, se ha descrito numerosas alteraciones, en pacientes con anemia aplásica asociada con virus, tales como la presencia de poblaciones celulares con carácter supresor, inversión del cociente CD4/CD8 y niveles elevados de IL-2, (interleukina-2), INF-gamma (interferón-gamma), y TNF, (factor de necrosis tumoral), sin embargo estos datos no son definitivos, pero si se ha logrado identificar el virus dentro de la célula (10). Mielopatía monoclonal La posibilidad de una mielopatía monoclonal selectiva para la célula germinativa, puede ser considerada desde que hay relación entre anemia aplásica y hemoglobinuria paróxística nocturna (HPN), en muchos casos de HPN, a lo largo de su evolución se convierten en anemia aplásica y otros casos de anemia aplásica desarrolla HPN (11). También está perfectamente establecido que a lo largo de su evolución la anemia aplásica, pueden desarrollar leucemia aguda mieloblástica o mielodisplasica. Trastornos de la regulación de la hematopoyesis La alteración de la capacidad funcional de las células del compartimiento accesorio (comprenden diferentes sobpoblaciones de linfocitos T, linfocitos B, células NK y monocitos) por un probable mecanismo inmunológico, corresponde a uno de los aspectos de la anemia aplásica; donde más se ha investigado. El desarrollo de un mecanismo inmune capaz de producir este tipo de lesión de la hematopoyesis, puede considerarse como el más probable. Desde el punto de vista biológico, se han estudiado los posibles mecanismos celulares y humorales capaces de desarrollar la supresión de la hematopoyesis, por ejemplo los linfocitos de pacientes transfundidos y de algunos no transfundidos puedes tener capacidad para inhibir el crecimiento de células germinativas (12). Se ha descrito una población celular con actividad supresora y con capacidad de sintetizar IFN-gamma (interferón gamma) en la producción de anemia aplásica, también se ha detallado un incremento del número de linfocitos activados (Tac +, HLA-DR +, CDS +) con niveles elevados de IIFN-gamma, en el suero de 10 de 24 pacientes con anemia aplásica, la producción espontánea de IFN-gamma, por parte de las células mononucleares de la sangre periférica, se encontraba elevado en estos pacientes y el número de colonias mieloides en el cultivo de médula ósea, se elevaba en estos pacientes y el número de colonias mieloides en el cultivo de médula ósea, se elevaba en estos pacientes tras la adicción de anti-IFN-gamma (13). Algunos autores opinan que las alteraciones descritas en las subpoblaciones linfocitarias. son consecuencia de la sensibilización producida por las transfusiones previas y no que correspondan a un mecanismo primario implicado en la producción de anemia aplásica (14). Otros trabajos han estudiado un posible compromiso de otras linfoquinas en la anemia aplásica, por que los niveles de IL-l se hallan disminuidos, mientras que los de IL-2 y el Factor de necrosis tumoral (TNF), se encuentran elevados (15). Como es conocido las células NK (matador natural), son las principales productoras. de estas linfoquinas en los pacientes con anemia aplásica, el número de células NK no es elevado, pero si el de la células pre-NK (16). Anemia aplásica hereditaria La causa más común de la anemia aplásica constitucional, es la descrita en tres hermanos por Fanconi en 1927, desde entonces se han descrito cerca de 800 casos. Esta anemia se hereda como una condición autonómica recesiva, a menudo ocho mutaciones genéticas han sido asociadas con la anemia de Fanconi. Los recuentos celulares y la celularidad de la médula ósea, es a menudo normal hasta la edad de 5 a 10 años, esta enfermedad es asociada con pigmentación anormal de la piel, retardo del crecimiento, displasia de la tibia y el pulgar. Microcefalia y retardo mental pueden existir. La otra forma hereditaria es la Diskeratosis Congénita y el Síndrome de Schwachman Diamond, caracterizado por insuficiencia pancreática, pudiendo comprometerse con anemia aplásica. La Diskeratosis es usualmente heredada como recesiva ligada al cromosoma X teniendo un progresivo riesgo de transformarse en leucemia mieloblástica. Epidemíología La incidencia de anemia aplásica, es estimada en 2 a 5 casos por /millón/año de habitantes Por ejemplo la incidencia en Suecia es de 13/millón/año, Israel 6/millón/año, USA es de 5 a 12 /millón/año, sugiriendo que el grado de incidencia es en los países industrializados es de cerca de 5 a 10/millón/año (8). El grado de incidencia de la anemia aplásica, fue establecido en 2/millón/año, por el estudio internacional de anemia aplásica y agranulocitosis (IAAAS). Estudio que fue conducido en Europa e Israel pero señalando diferencias geográficas. Los grados de anemia aplásica, fueron inversamente proporcionales a los de la agranulocitosis. El IAAAS, también cuantificó el riesgo absoluto y relativo para un gran número de drogas de uso médico, encontrándose la fracción etiológica para la anemia aplásica, del 25% y 65% para la agranulocitosis (17). Cuadro clínico El inicio puede ser insidioso, con descenso gradual de los niveles de hemoglobina, palidez debilidad y fatiga, en otros se suman procesos infecciosos como resultante de la neutropenia y hemorragias como consecuencia de la trombocitopenia. Se pueden considerar dos tipos clínicos: una de anemia aplásica de evolución más lenta (AA) y otra la anemia aplásica severa (AAS), el segundo está caracterizado por una acentuada disminución de los neutrófilos < 0,5x10/9/ L, lo que le concede el peor pronóstico, en cambio el primero con mejores niveles de leucocitos le da una evolución clínica más larvada (18). Al examen clínico nos encontramos con un paciente que presenta palidez, sangrado, fiebre y sin esplenomegalia. Hallazgos de laboratorio En los hallazgos de laboratorio, los grados de pancitopenia varían, pero el nivel de reticulocitos generalmente es menor del 1%. Puede encontrarse macrocitosis, como resultado de los altos niveles de eritropoyetina presentes en el plasma de estos pacientes, lo que estimula a los eritroblastos residuales a madurar más rápidamente (19). Cuando los leucocitos son < 0.5x10/9/L; las plaquetas < 20x10/9/L; y los reticulocitos < del 1% , estos hallazgos le conceden peor pronóstico y cataloga como anemia aplásica severa (ASS). Hallazgos de médula ósea El aspirado de médula ósea, demuestra espículas con celularidad muy disminuida e incremento del tejido graso. Los linfocitos y células plasmáticas pueden observarse incrementados, en otras ocasiones algunas espículas conservan su celularidad o aun pueden aparecer hipercelulares, pero los megacariocitos invariablemente están disminuídos. La AAS ha sido definida por International Aplastic Anemia Study Group (20) en las siguientes categorías en relación a la MO; de menos del 25 % de celularidad, con menos del 30 % de células hematopoyéticas; o menos del 50 % de celularidad con menos del 30 % de células hematopoyéticas; con al menos dos de los siguientes hallazgos: granulocitos menos de 500 xmm3, plaquetas menos de 20,000 xmm3 y reticulocitos, menos del 1% (corregidos por hematocrito). Tratamiento La sobrevida y las posibilidades de curación de los pacientes con AA, han mejorado en forma ostensible en las últimas décadas, coincidiendo con los avances terapéuticos con drogas inmunoestimuladoras y transplante de médula ósea.
Aquellos pacientes con enfermedad menos severa, con neutrófilos encima de 1,000 xmm3, que no tienen grandes requerimientos de sangre o plaquetas, pueden ser tratados conservadoramente con medidas de soporte, pudiendo emplearse derivados androgénicos y glucocorticoides, que tienen alguna capacidad para estimular la MO. Inclusive pueden encontrarse casos de curación espontánea (21). Transplante de médula ósea El TMO, es una terapia agresiva, que está indicada en la mayoría de pacientes con (AAS), obteniéndose los mejores resultados con transplantes alogénicos. La intención es hacer el reemplazo de la hematopoyesis alterada, con células de MO normal de donante HLA idéntico. El TMO es el único tratamiento que ha demostrado poder curar a largo plazo la AAS, pero también hay que considerar que hay una serie de complicaciones severas como consecuencia del tratamiento (22). Considerándose dentro de ellas, como las más importantes. el rechazo del injerto, la enfermedad del injerto contra el huésped (EICH) y las complicaciones infecciosas. En el TMO, de gemelos hemocigóticos es posible en 50%, transfundir MO sin condicionamiento previo, pero el 50% restante, es necesario el condicionamiento. Los primeros resultados obtenidos en los años de la década del setenta, tenían un promedio de rechazo del 30% al 40%, en pacientes con AAS, condicionados exclusivamente con ciclofosfamida (23). Existe una serie de variables, que inciden directamente en el rechazo, por ejemplo el número de transfusiones recibidas, una cantidad de células nucleadas inferior a 3x10/8/L. Se ha utilizado la administración, en la fase post transplante, de la capa leucocitaria y plaquetería del donante, en el intento de facilitar el injerto con las células del donante (24) , pero se incrementó el EICH crónico. En pacientes no transfundidos y condicionados previamente con ciclofosfamida y utilizando profilaxis del EICH, con metrotexato y ciclosporina, el rechazo aparece en menos del 5% (25). Otros grupos han asociado radioterapia, obteniendo una incidencia de rechazo entre 5 a 15%. Los pacientes con AAS, trasplantados, los resultados varían entre 40 a 80% (26), donde es posible demostrar la gran influencia de la menor edad, así los pacientes entre 10 y 20 años, la sobrevida puede superar el 80%, en los casos mayores de 20 años varían entre 40 y 60%. Por tal razón los pacientes con ASS, son considerados desde el diagnóstico candidatos al TMO. Tratamiento inmunoestimulador Las primeras experiencias en este terreno fueron realizadas por Mathé (27) pero no se establecieron hasta los primeros resultados presentados por Speck (28) quien demostró una recuperación de la hematopoyesis en alrededor del 40 % de los pacientes tratados. Trabajos posteriores han confirmado estos resultados preliminares. En estudios controlados, la Globulina Antitimocítica (GAT), ha demostrado ser superior a las terapias convencionales de, soporte(29). Las complicaciones más frecuentes del tratamiento con GAT, son fiebre, rash cutáneo, trombocitopenia y enfermedad del suero. Después de la administración de GAT, la respuesta cuando la hay, no se observa antes de los dos a tres primeros meses. Se obtiene respuesta completa alrededor del 20 % de casos y una respuesta parcial con disminución o desaparición de los requerimientos transfusionales, pero sin normalización de los recuentos periféricos en un 30 a 40 %. Alrededor del 15 a 20 % de los pacientes pueden recaer, pasado uno o dos años, pero tienen opción a un nuevo tratamiento, pero no hay ningún dato que pueda predecir la buena respuesta (30). Parece ser qué, aquellos pacientes con un intervalo de tiempo menor entre el diagnóstico y el tratamiento, responden mejor que los que presentan una evolución más prolongada. La respuesta no está influenciada por la edad, como lo está es el caso del TMO. Existen varios trabajos que han comparado los resultados del TMO y el GAT. El estudio más amplio es el del Grupo Europeo de transplante de MO, publicado por Bacigalupo (31). De los análisis de sus resultados se puede se deducir, que en pacientes por debajo de los 20 años y con granulocitos inferiores a 200 xmm3, el TMO se presenta como el más eficaz que la GAT (64% versus 38%). Sin embargo si la cifra de granulocitos es superior a 200 xmm3, los resultados son similares (62.5 versus 58%). Bibliografía
(*) Presidente de la Sociedad Peruana de Hematología. Profesor Principal de la Universidad Nacional Mayor de San Marcos (UNMSM). |
|||