SÍNDROMES ANÉMICOS
(Segunda Parte)
Anemias hemolíticas congénitas
Dr. Oscar Ruiz Franco (*)
Las anemias hemolíticas congénitas (intracorpusculares) se definen como aquellas en donde el glóbulo rojo, en alguna parte de su estructura presenta alteraciones. Se pueden dividir en tres grandes grupos: membranopatías, enzimopatías y hemoglobinopatías (Tabla 1) (1,2).
Independientemente de la causa de la anemia hemolítica congénita, en este grupo de entidades no se recomienda la administración de hierro dado que los depósitos de éste oligoelemento se encuentran incrementados, e inclusive existe el riesgo de condicionar el desarrollo de hemocromatosis. El uso de paquete globular se debe indicar según la fisiopatología de cada entidad (1,2).
Anemias Hemolíticas Congénitas
1 Membranopatías.
• Esferocitosis hereditaria.
• Eliptocitosis congénita.
• Estomatocitosis congénita.
• Acantocitosis congénita (Abetalipoproteinemias)
2. Enzimopatías.
• Deficiencia de glucosa-6-fosfato deshidrogenasa.
• Déficit de piruvato kinasa
• Otras enzimopatías.
3. Hemoglobinopatías.
• Hemoglobinopatías estructurales (Hemoglobinas anormales)
• Talasemias
1. Membranopatías
Las membranopatías obedecen a defectos en alguno de los componentes estructurales de la membrana. Las de mayor interés clínico son: esferocitosis hereditaria (EH), eliptocitosis congénita (EC), estomatocitosis y acantocitosis (2,3).
1.1 Esferocitosis Hereditaria
Se conoce también con el nombre de Minkowsky Chauffard. Es la anemia hemolítica más frecuente en la raza blanca. En el 80% de los casos, se transmite con carácter autosómico dominante. Se caracteriza por su escasa expresividad clínica, la cual consiste en anemia moderada con esferocitosis y respuesta completa a la esplenectomia (2,4,5).
Fisiopatología
La causa es la deficiencia o anormalidad de una o más proteínas de la membrana del eritrocito, por lo que aparece: liberación de lípidos, menor área de superficie del eritrocito y formación de esferocitos poco elásticos. La mayoría de pacientes presenta una deficiencia combinada de espectrina y anquirina. En la deficiencia de espectrina se pierde lípidos, pues la bicapa lipídica no tiene el sostén del esqueleto (2,5,6).
La disminución en la proporción superficie/volumen y el aumento en la viscosidad interna, vuelven a los esferocitos menos deformables al pasar por los senos esplénicos, por lo tanto son destruidos. Tanto la pérdida de la membrana como la deshidratación del eritrocito, producen un incremento en la concentración de hemoglobina corpuscular media (CHCM), la cual usualmente es mayor a 35 mg/ dL (2,5,6).
Manifestaciones Clínicas
La sintomatología aparece durante las primeras décadas de la vida y rara vez en la edad adulta. Las formas clínicas se clasifican según la intensidad de la anemia, siendo la más frecuente, la forma moderada la cual se observa entre el 60 a 65% de casos. Esta forma moderada se caracteriza por anemia discreta, ligera esplenomegalia e ictericia intermitente (6).
| TABLA 1 |
| MECANISMOS MOLECULARES DE LA ESFEROCITOSIS HEREDITARIA (2) |
|
|
| 1. Deficiencia de anquirina Frecuencia Mecanismo molecular Expresividad |
40 - 50%
Defecto de la transcripción.
Mutaciones en región promotora
Esterocitos y acidosis tubular |
| 2. Deficiencia de Banda 3
Frecuencia
Mecanismo molecular
Expresividad |
20 - 30%
Mutación de fragmento citoplasmático.
Esterocitos y acidosis tubular |
| 3. Deficiencia de espectrina Frecuencia Mecanismo molecular Expresividad |
10 - 15%
Mutación de codón de iniciación.
Esquinocitos y acantocitos |
| 4. Deficiencia de espectrina
Frecuencia
Mecanismo molecular
Expresividad |
5 - 10%
Mutación puntual / gen LEPRA*
ESFEROCITOS INTENSA |
| 5. Deficiencia de proteína 4,2
Frecuencia
Mecanismo molecular
Expresividad |
Frecuente en Japón.
Mutaciones puntuales.
Esferocitos. |
|
Diagnóstico
El diagnóstico de EH no siempre es fácil y se basa sobretodo en la morfología eritrocitaria, estudio de fragilidad osmótica y estudio familiar. En el hemograma se destaca la presencia de microesferocitos cuyo porcentaje puede variar desde 3% a 30%. Las constantes corpusculares muestran una disminución del volumen corpuscular medio (VCM) y un incremento significativo de la CHCM mayor de 35 mg/ dL (2,6).
| figura 1 |
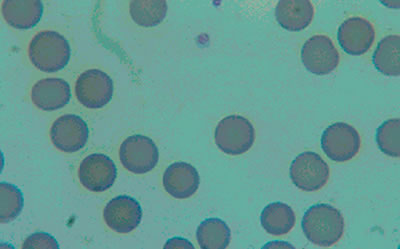 |
| Figura 1. Lámina periférica de un paciente con esferocitosis hereditaria. Obsérvese la presencia de abundantes esferocitos |
Prueba de Fragilidad Osmótica: Consiste en someter los glóbulos rojos de los pacientes a medios hipotónicos. Se practica a la hora y a las 24 horas luego de haber sido extraída la muestra. En condiciones normales los eritrocitos sin incubar (prueba inmediata) inician la hemólisis a la concentración de 5,0 gr/dL de suero fisiológico y es total a 3,5 gr/ dL, mientras que los eritrocitos incubados (prueba de 24 horas), presentan valores de 6,0 gr/ dL y 4,0 g/ dL, respectivamente. La prueba incubada es la más sensible y es positiva en el 100% de esferocitosis hereditaria (2,6).
Tratamiento
El tratamiento de elección es la esplenectomia pues el objetivo es eliminar el lugar donde se destruyen los eritrocitos. Una condición importante es tener en cuenta la edad, ya que en niños, no es aconsejable practicarla salvo que haya un requerimiento transfucional alto o si hay signos de retraso en el crecimiento óseo y corporal. En cualquier caso se debe practicar después de los 5 ó 6 años. Se deben vacunar a los pacientes esplenectomizados contra el neumococo, H. influenza tipo b y meningococo. Además se recomienda la administración de folato a dosis de 1 mg/ día para evitar las crisis megaloblásticas (2,3,6).
1.2 Eliptocitosis Congénita
Es una membranopatía que tiene como patrón característico la presencia de glóbulos rojos ovalado o elipticos, hallazgo que hasta la fecha es el principal criterio diagnóstico. Se transmite por herencia autosómica dominante y su frecuencia es de 1 x 5 000 RNV. La enfermedad tiene un notable polimorfismo clínico y molecular. Las mutaciones causantes del cuadro se pueden hallar en tres genes diferentes: 1 espectrina, espectrina y proteína 4,1(1,2).
Manifestaciones Clínicas
La eliptocitosis congénita se puede clasificar en cuatro grupos:
a. Eliptocitosis congénita común.
b. Piropoiquilocitosis congénita.
c. Eliptocitosis congénita esferocítica.
d. Eliptocitosis congénita estomatocítica. A continuación se describe las características más saltantes de la Eliptocitosis congénita común. Se trata de la variante más frecuente y sus manifestaciones pueden presentar diferente grado de intensidad. Así pues, hay personas afectadas que no presentan síntomas hasta aquellas que expresan un cuadro hemolítico intenso. Este último, se caracteriza por una marcada anemia con esplenomegalia y laboratorialmente, en el frotis de sangre periférica se observan eliptocitos, ovalocitos y dacriocitos (1,2).
Diagnóstico
En el hemograma se encuentra la presencia de eliptocitos (eritrocitos con relación diámetro longitudinal/ transversal > 1) en porcentajes mayores al 12%. El diagnóstico definitivo se hace con el análisis del ADN para identificar el tipo de mutación (1,2).
Tratamiento
La esplenectomía sigue siendo el único tratamiento eficaz aún cuando la respuesta no es tan completa como en la esferocitosis hereditaria (1,2).
1.3 Estomatocitosis Congénita
Corresponde a un grupo heterogéneo de trastornos de la membrana del glóbulo rojo, cuya característica es un defecto de la permeabilidad a los iones monovalentes (sodio y potasio).
En la actualidad se reconocen cinco síndromes estomatocíticos congénitos con transmisión hereditaria autosómica dominante: síndrome Rh0, hidrocitosis congénita, xerocitosis congénita, pseudo hiperpotasemia hereditaria y ovaloestomatocitosis asiática. Todos ellos presentan un cuadro clínico de anemia hemolítica crónica de intensidad variable (1,2).
2. Enzimopatías
Se han descrito numerosas enzimopatías que comprometen diferentes vías del metabolismo eritrocitario, para su estudio, estas entidades se pueden agrupar en 3 grupos (1,2,3).
• Enzimopatías de la glucolisis anaerobia.
• Enzimopatías del metabolismo oxido reductor.
• Enzimopatías del metabolismo nucleotídico.
2.1 Deficiencia de Glucosa 6
Fosfato Deshidrogenasa
Fue la primera en describirse como causas de hemólisis enzimática. Esta enzima cataliza la primera reacción de la vía de las pentosas fosfato y su función principal es proteger el eritrocito de agentes oxidantes. Es un trastorno ligado a herencia sexual (cromosoma X) y constituyéndose en la alteración mejor conocida. Tiene mayor predilección por personas de raza negra, blancos de la zona del Mediterráneo y asiáticos. La deficiencia de G-6-P-D en personas de raza blanca es grave, pues la actividad enzimática en ellos es apenas detectable. La variedad G-6-P-D “A”(-) muestra sólo una actividad enzimática de 5-15%, por lo que ocasiona un cuadro hemolítico severo (1,2,3).
Desencadenantes de Hemólisis
• Fármacos Oxidantes como son sulfas, nitrofuranos, furazolidona, fenazopiridina, primaquina, fenilhidrazina.
• Procesos febriles: neumonías, fiebre tifoidea, septicemia, etc.
Cuadro Clínico
Consiste en la presencia de un síndrome hemolítico crónico de intensidad variable. Un cuadro clínico característico y frecuente es el favismo (por consumo de habas), el cuál es un episodio de hemólisis aguda , siendo la población mas susceptible los niños menores de 5 años. El agente desencadenante es la divicina, el cual es un derivado de las habas, que posee un fuerte efecto oxidante. El cuadro hemolítico agudo consiste en una anemia hemolítica intensa, de aparición brusca, acompañado de coluria, cefalea, fiebre e ictericia (1,2,3).
Diagnóstico
Consiste en medir la actividad enzimática y establecer el tipo de mutación (2).
Tratamiento
Luego de establecer el diagnóstico se debe evitar el contacto con sustancias capaces de desencadenar hemólisis. La esplenectomía solo se indicara en cuadros hemolíticos que comprometan la vida de la persona. En los cuados de hemólisis crónica se recomienda la administración de ácido fólico 1 mg/ día. (2,3,4).
2.2 Deficiencia de Piruvato Kinasa (PK).
Es la enzimopatía de la glucólisis anaeróbica más frecuente. La piruvato kinasa es la última enzima de la glucólisis y cataliza la transformación de fosfo-enol-piruvato a piruvato, proceso en que se produce una molécula de ATP. La PK humana es codificada por dos genes diferentes (PK-RL y PK-M). La deficiencia congénita de la PK eritrocitaria se transmite con carácter autonómico recesivo y hasta la fecha se han descrito algo más de 400 casos en todo el mundo (1,2).
Manifestaciones Clínicas
La deficiencia de PK cursa con anemia hemolítica de intensidad variable. El cuadro hemolítico se puede presentar desde el periodo neonatal o en la primera década de la vida y sus características son parecidas a las del cuadro clínico de la esferocitosis hereditaria, excepto por la presencia de esferocitos circulantes y fragilidad osmótica normal (1,2).
Diagnóstico
La deficiencia de PK requiere siempre de la medida de la actividad enzimática en el hemolisado. Se debe tomar la precaución de eliminar por completo los leucocitos ya que estas células poseen una actividad PK normal y podrían enmascarar la carencia de la PK en los eritrocitos (1,2,10).
Tratamiento
El tratamiento más efectivo sigue siendo la esplenectomía, aunque su éxito es inferior al observado en la esferocitosis hereditaria. Se recomienda el uso de ácido fólico para evitar el agotamiento de las reservas y aparición de una crisis megaloblástica.
3. Hemoglobinopatías
Las alteraciones congénitas de la hemoglobina se clasifican en dos grandes grupos: defectos estructurales de la hemoglobina (hemoglobinopatía S) y disminución de la síntesis de hemoglobina (talasemias)(2,3). .
3.1 Anemia Drepanocitica (Hemoglobinopatía S)
Incidencia y Distribución Geográfica
La mayor incidencia corresponde a la región de África Tropical, donde hasta un 45% de la población es portadora de la mutación. Otra área importante es la zona adyacente al Mediterráneo. En América, se presenta con frecuencia en los países con población negra significativa: Estados Unidos, Brasil y países del Caribe. En el Perú, se han descrito como lugares de alta incidencia las costas de Ica y Lima, debido a que la presencia de población negra es significativa (2,3,7,8).
Etiopatogenia
Se debe a la sustitución del ácido glutámico en posición 6 de la cadena beta por valina, con lo cual hay sustitución de adenina por timina en el código ADN. Las moléculas de desoxihemoglobina S se agregan ordenadamente, formando microtúbulos en forma helicoidal; debido a ésto surge la forma de hoz del eritrocito (7,8).
Clínica
Existen dos formas clínicas de HbS: homocigota (HbSS) y heterocigota (HbAS). La más frecuente es la HbAS, llamada rasgo drepanocítico (8,9).
Anemia falciforme (HbSS)
En esta entidad la intensidad de la hemólisis varía de un paciente a otro y su inicio, debido al efecto protector de la hemoglobina fetal (HbF) es siempre después de los 4 a 6 meses de vida. En el desarrollo clínico se pueden considerara tres fases: estacionaria, de expresividad aguda y de expresividad crónica (2,8,9,10).
Fase Estacionaria: Corresponde a los primeros años de vida (1-4 años). Tiene manifestaciones clínicas de un cuadro hemolítico moderado o intenso: anemia, ictericia y retraso del crecimiento óseo y gonadal. Es característica la presencia de crisis vaso oclusivas que conducen a la pérdida de la función esplénica o auto esplenectomia (2,10).
Fase de Expresividad Aguda: A partir de los 4 años con agravamiento de la anemia (hemoglobina menor de 8 gr/ L). Las crisis vaso oclusivas afectan al pulmón, riñón y tejido óseo; caracterizándose por dolor intenso en los territorios afectados (2,10).
Crisis de Dolor Agudo: El dolor óseo limitado o generalizado es lo más característico y obedece a oclusiones de la micro vasculatura. Las infecciones son la complicación más frecuente de este cuadro siendo las responsables de la mayoría de muertes. Su incidencia va disminuyendo con la edad. Las infecciones más frecuentes son debidas a neumococo y H. influenza tipo b (2,10).
El cuadro llamado síndrome toráxico, consiste en la presencia de fiebre, taquipnea y dolor toráxico intenso, es la causa más frecuente de hospitalización, pudiendo ser causa de muerte. Otros territorios comprometidos pueden ser la retina, el sistema nervioso central y los cuerpos cavernosos del pene (2,10).
Fase de Expresividad Crónica: Se presenta en la adolescencia y en la edad adulta, afectando el crecimiento y el desarrollo corporal así como el sistema nervioso central, cardiovascular, pulmonar, gastro intestinal y renal. Otra complicación relativamente frecuente en esta fase es la presencia de úlceras maleolares de evolución tórpida (2,3,8).
| TABLA 2 |
| MANIFESTACIONES CRÓNICAS
DE LA ANEMIA FALCIFORME (2) |
|
• Úlceras maleolares
• Necrósis óseas (enfermedades de Legg-Calvé-Perthes)
• Complicaciones visuales (retinopatía)
• Complicaciones pulmonares (insuficiencia respiratoria)
• Complicaciones cardiacas (insuficiencia cardiaca)
• Complicacioneas renales (hipostenuria)
• Complicaciones hepatobiliares (cirrosos difusa nodular)
• Complicaciones debidas a hiperbilirrubinemia |
|
Rasgo Falciforme (HbAS)
Este rasgo corresponde a los portadores heterocigotos del gen falciforme y se caracteriza por la ausencia de signos clínicos. La hemoglobinopatía se pone de manifiesto mediante pruebas de laboratorio (electroforesis de hemoglobina) (8,9).
La enfermedad se manifiesta en situaciones de estrés con disminución de la presión parcial de oxígeno: altitud, submarinismo, anestesia o luego de realizar un esfuerzo físico intenso. El cuadro clínico en estas personas se refiere a las crisis vaso oclusivas que comprometen sobretodo el bazo y el riñón (8,9).
Diagnóstico
Hemograma: El hemograma muestra una anemia normocítica o ligeramente macrocítica con hemoglobina entre 7 y 9 gr/ dL, acompañada de reticulocitosis. El frotis evidencia la presencia de drepanocitos y dianocitos (2,9).
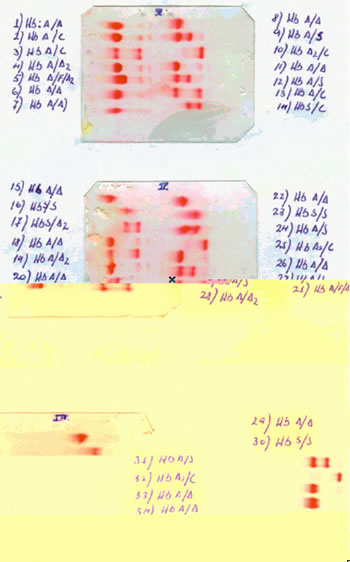
Electroforesis de Hemoglobinas a pH alcalino: Es el procedimiento diagnóstico más usado. Los individuos homocigotos HbSS muestran una fracción de hemoglobina que migra por detrás de la hemoglobina fetal (HbS) y ausencia total de hemoglobina A. Los sujetos heterocigotos HbAS, presentan dos fracciones hemoglobinicas de intensidad similar que corresponden a la hemoglobina A y S. En ambas situaciones, la hemoglobina A2 es normal. Pueden existir doble heterocigotos, como por ejemplo rasgo falciforme asociado a rasgo talasémico (HbAS + Talasemia) (2,9).
| figura 2 |
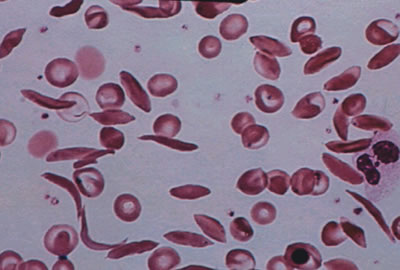 |
| Figura 2: Lámina periférica de un paciente con drepanocitosis. Obsérvese la presencia de abundantes drepanocitos (células alargadas que simulan la forma de un plátano). |
Tratamiento
Tratamiento preventivo: Evitar todas aquellas situaciones que desencadenen las crisis vaso oclusivas: infecciones, acidosis, hipoxemia y exposición al frío (2,9,10).
Tratamiento curativo:
Folato: 1 - 5 mg/ día en forma permanente.
Transfusiones sanguíneas: solo cuando sea necesario, pues al aumentar la viscosidad sanguínea se incrementa el riesgo de hemólisis. Los pacientes con anemia falciforme presentan una gran adaptación a la anemia, llegando inclusive a tolerar límites tan bajos como 5 gr/ dL. En caso de ser necesaria la transfusión, no se debe exceder como objetivo 10 gr/ dL (2,10).
Hidratación adecuada: cuyo objetivo es evitar la hiperviscosidad sanguínea, para lo cual se recomienda cantidades que varíen entre 2 y 4 L/ m2 por día (2,10).
Analgésicos: de acuerdo a la intensidad del dolor.
Agentes antidrepanociticos: Hidroxiurea, cuyo objetivo es incrementar la concentración de hemoglobina fetal (HbF), disminuyendo por lo tanto la frecuencia de las crisis vaso oclusivas. Se recomiendan dosis de 15 -20 mg/ Kg/ día (2,10).
Transplante de Médula Ósea: Este procedimiento se debe reservar para los casos de mal pronóstico o incompatibles con una calidad de vida mínimamente aceptables. Se debe realizar en pacientes menores de 16 años con donante compatible. Los datos internacionales describen un éxito de hasta el 80% (2,9,10).
| figura 3 |
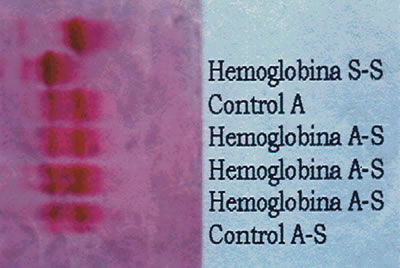 |
| Figura 3. Electroforesis de Hemoglobinas. Obsérvese el patrón de migración diferente del paciente portador de
Hemoglobina SS respecto del control sano A. |
3.2 TALASEMIAS
Las talasemias son entidades en las que existe una alteración cuantitativa de las cadenas que forman la porción globina de la hemoglobina. Pueden verse afectadas tanto las cadenas alfa como las no alfa. Si bien las alfa talasemias son las más frecuentes, las beta talasemias son las mejor estudiadas (1, 2, 3).
Beta Talasemia Mayor (Enfermedad de Cooley)
La forma más representativa es el estado homocigoto ( °/ °), que se caracteriza por un predominio casi absoluto de hemoglobina fetal y ausencia de hemoglobina A (el patrón hemoglobinico no presenta cadenas ), la cual es debida a una mutación puntiforme; esto se expresa como microcitosis e hipocromía. Su presentación es mas frecuente en la raza negra y en la población blanca del Mediterráneo. Dentro de sus variantes tenemos el homocigoto ( +/ +) y los doble heterocigotos ( °/ +) (1,2,3).
Cuadro Clínico
La anemia se inicia a partir del sexto mes de nacimiento, siendo ésta de intensidad grave lo que obliga a recurrir a transfusiones periódicas. Generalmente se acompaña de esplenomegalia que aparece al tercer año de vida y de una hepatomegalia variable. También son hallazgos característicos las alteraciones óseas, especialmente en el cráneo y la cara, implantación anómala de dientes y alteraciones en la configuración normal del organismo (2,3).
Diagnóstico
Hemograma: Se caracteriza por una anemia severa (hemoglobina < 7,0 gr/ dL) de carácter microcitico e hipocrómico. El frotis periférico evidencia una marcada microcitosis, punteado basófilo, dianocitos y presencia de glóbulos rojos nucleados (ortocromáticos). Se puede observar leucocitosis y trombocitosis, sobre todo después de realizada la esplenectomía. Los reticulocitos se hallan incrementados y su elevación guarda relación con el grado de anemia (2,9).
Mielograma: Los depósitos de hierro se encuentran incrementados y la celularidad hematopoyética se caracteriza por ser hiperplásica sobretodo en la serie eitroide (2,9).
Electroforesis de Hemoglobinas: Evidencia un incremento de la hemoglobina fetal entre 60% y 98%. La hemoglobina A se halla ausente y la hemoglobina A2, se encuentra dentro de límites normales (2,9).
Tratamiento
Transfusiones sanguíneas: Tienen como objetivo mantener los niveles de hemoglobina alrededor de 10,0 g/ dL para asegurar un desarrollo psico motriz normal y una mejor calidad de vida del paciente. Es mejor transfundir glóbulos rojos recién extraídos (sangre fresca) (2,10,11).
Quelantes de Hierro: La deferoxamina es el más usado y menos tóxico. Este fármaco se administra por vía subcutánea a dosis de 50 a 60 mg/ Kg (9).
Esplenectomía: Se recomienda en los pacientes con intensa esplenomegalia sobretodo si hay compresión de órganos vecinos e hiperesplenismo. Muchas veces, esto contribuye a la disminución del requerimiento transfusional (9).
Transplante de Médula Ósea: El transplante medular alogénico se utiliza con mucho éxito desde 1982 y los mejores resultados los ha mostrado el grupo del Profesor Lucarelli de la Universidad de Pesaro (superviviencia promedio libre de enfermedad a 5 años 90%) (9).
Beta Talasemia Menor
(Heterocigoto o Rasgo Talasémico)
Corresponde a los estados heterocigotos para los genes de la cadena beta, en donde una de ellas se encuentra alterada mientras que la otra, es normal ( +/ y °/ ). El cuadro clínico se caracteriza por la presencia de anemia muy leve y en muy pocos casos se puede presentar esplenomegalia (2,3,9).
Diagnóstico
Hemograma: Se caracteriza por la presencia de anemia leve de tipo microcítica hipocrómica con reticulocitosis. En el frotis periférico se puede encontrar la presencia de dianocitos y punteado basófilo (2,6,8,9).
Médula ósea: De manera semejante a lo descrito en Beta Talasemia mayor, se suele encontrar aumento de la hemosiderina y maduración megaloblástica por consumo de folato, pero en menor intensidad que lo descrito para el primer caso (2,8,9).
Electroforesis de Hemoglobina: Característicamente se presenta un incremento de la hemoglobina A2, mientras que la hemoglobina fetal puede estar normal o discretamente elevada (2,8,9).
Tratamiento
Su objetivo es básicamente preventivo y se consigue mediante la administración de ácido fólico a dosis de 1 mg por día. No se debe administrar suplementos de hierro por el riesgo de hemocromatosis (2).
Bibliografia
- Lee et al. “Wintrobe's Clinical Hematology”. 10 ma Edición. Williams & Wilkins. Houston. 1999.
- Sans Sabrafen y col. “Hematología clínica”. 4ta Edición. Editorial Hartcourt. Madrid España. 2001.
- Ernest Beuler et al. "Williams Hematology". 5ta Edición. Editorial Mc Graw - Hill. 1995.
- Weiner, M. et al. "Pediatric Hematology/ Oncology Secrets". 2 da Edición. Editorial Hanley & Belfus. 2002.
- Wood M. y col. "Secretos de la Hematología y Oncología". 2da Edición. Editorial Interamericana. 2000.
- Dacie, J. "The haemolytic anemias" 3ra Edición. Editorial Churchill & Livingstone. 1985.
- Ruiz Franco y col. "Estudio de Hemoglobinas anormales en una Población de Raza Negra en el Perú". Sangre. 1990;35: 263-5.
- Castilo y col. "Las Hemoglobinopatías en el Perú". Rev. Médica del IPSS. 1998;7 (1) :7 - 17.
- Ruiz, Franco "Hemoglobinopatías en una Muestra de Pobladores de Raza Negra en el Distrito de La Victoria - Lima". Tesis de Maestría en Fisiología. UNMSM. 2001.
- Mentzer, WC. "The hereditary hemolytic anemias". 1ra Edición. Editorial Churchill & Livingstone. 1989.
- Hebbel, RP. "Beyond hemoglobin polymerization: the red blood cell and sickle cell disease pathophysiology". Blood. 1991;2:214 - 37.
(*) Profesor Asociado Facultad de Medicina Universidad Nacional Mayor de San Marcos (UNMSM).
Médico Asistente del Hospital Nacional “Dos de Mayo”. |