Estudio clínico molecular de la Enfermedad de Huntington
en pacientes del Valle de Cañete - Perú
Luis Torres Ramírez (1), Nicanor Mori Quispe (2), Marleni Mendoza Cabanillas (2), Jorge Montoya Henriquez (2),
Mariano Cuentas Jara (2), José Domínguez Calderón (2), Hilda Mendoza Chávez (2), David Pérez Díaz (2), Pilar Mazzetti Soler (3)
Resumen
Objetivos:Estudiar las características clínicas y moleculares de 30 pacientes con Enfermedad de Huntington (EH) provenientes del Valle de Cañete. Población y métodos: Estudio analítico, transversal. Se estudió el ADN de 30 pacientes para determinar el número de tripletes CAG. Resultados: De 30 pacientes, 17 (56.7%) fueron varones, 13 (43.3%) mujeres, con edades entre 23 y 71 años y una media de 43.2 años. La manifestación clínica de inicio de la enfermedad más frecuente fue la corea. La expansión dentro del límite patológico del triplete CAG de 20 pacientes osciló entre 40 y 56 repeticiones mientras que en los alelos normales estuvo comprendido entre 14 y 33. Diez pacientes presentaron rangos entre 36 y 39 tripletes. Un paciente con 43 repeticiones inició la enfermedad antes de los 20 años. Se halló correlación inversa entre el número de tripletes CAG y la edad de inicio de la enfermedad (r = -0.718; p 0.001) y entre el Minimental Test de Folstein y el UHDRS (r = -0.678, p < 0.01) mientras que se halló correlación directa entre el UHDRS y el tiempo de enfermedad (r = 0.773, p < 0.001). Conclusiones: Aunque el número de repeticiones esta fuertemente correlacionado con la edad de inicio, estos datos no serán particularmente útiles en el diagnóstico presintomático excepto en aquellos pacientes con un alto número de repeticiones.
Palabras clave: Cañete, Enfermedad de Huntington, Huntingtina, Perú, Triplete CAG.
Abstract
Objetives: To study the clinical and molecular features of 30 patients with Huntington’s disease (HD) from 12 families of Cañete’s Valley. Population and methods: Analitic and transversal study, in wich the DNA of 30 patients was studied to derermine the CAG-repeats. Results: 30 patients 17 (56.7%) were male and 13 (43%) women. Overall, the mean age was 43.2 years with a range of 23 - 71 years. The first sympton of onset was chorea for the majority of patients. The molecular analysis revealed a range of 40 - 56 repeats in 20 affected individuals, and 10 between 36- 39 repeats. The normal chromosomes contained 14 to 33 repeat units. One patient with 43 CAG-repeats onset before 20 years age. We found a significantly negative correlation between the number og CAG-repeats and the age of onset (r = -0.718; p 0.001) and Folstein’s Minimental Test and UHDRS (r = -0.678, p < 0.01) while UHDRS and time of disease shown a positive correlation (r = 0.773, p < 0.001). Conclusions: Althoght repeat lenght is strong correlated with the age of onset, the data will not be particularly useful for presymptomatic testing except for those individuals who have very high repeat lenghts.
Key words:Cañete, Huntington’s Disease, Huntingtin, Perú, CAG Triplet.
Introducción
La Enfermedad de Huntington (EH) es un trastorno neurodegenerativo autosómico dominante (1, 2), se presenta en la adultez con movimientos involuntarios (3-5), trastornos psiquiátricos y deterioro de las funciones cognitivas (6-10), las cuales conducen a la muerte 15 a 20 años después del inicio de los síntomas (11). La EH ocurre con una frecuencia de 1 a 4 en 100 000 individuos en poblaciones caucásicas. La mayoría de los pacientes inicia la enfermedad en la cuarta década de la vida (12, 13). En un reducido número de pacientes se presenta antes de los 20 (EH juvenil) o después de los 50 años de edad (EH de inicio tardío) (14). Un 80% de pacientes que heredan la enfermedad por vía paterna la presentan en etapa juvenil, caracterizada por rigidez y akinesia, mas que por movimientos de tipo coreico (15).
En 1983 se descubre la alteración genética en el cromosoma 4p16.3 y 10 años más tarde se observa que la mutación se caracterizaba por aumento de repeticiones del triplete CAG, el cual codifica el aminoácido glutamina (16). En la EH, el gen afectado HD codifica la proteína huntingtina, con función aún desconocida, pero que posiblemente está implicada en el desarrollo embrionario normal, en la hematopoyesis y en la neurogénesis (17, 18). La EH es una de las 9 enfermedades hereditarias neurodegenerativas por aumento de tripletes CAG (19, 20), y es de todas la más prevalente. La huntingtina presenta normalmente hasta 35 residuos de glutamina en el extremo N-terminal, mientras que la forma mutante muestra 38 o más residuos y forma agregados cuya translocación hacia el núcleo parece ser un hecho crítico que induce la muerte neuronal por apoptosis (21). Se ha establecido que en los cromosomas normales el tamaño del triplete oscila entre 6 y 35 repeticiones, mientras que aquellos con más de 40 repeticiones desarrollarán siempre la enfermedad. Los pacientes con 36 y 39 repeticiones tienen un riesgo incrementado de desarrollar la enfermedad (19, 22).
En Perú Saavedra (23) en 1950, da a conocer el primer caso de EH en un paciente de 39 años, procedente de La Unión-Arequipa, con una historia de 9 parientes afectados. La enfermedad la heredó del padre, quien era proveniente de Cotahuasi-Arequipa. Cuba et al. (24) en 1983, presenta los primeros datos sobre la epidemiología de la EH en el Perú, llama la atención sobre la alta prevalencia en la provincia de Cañete-Lima. En 1986 (25) y 1989 (26), se realizaron los primeros estudios sobre genealogía de la EH identificando a Cañete como el principal foco de ésta enfermedad en Perú.
Este estudio es el primero que se realiza en el Perú con confirmación genética de la EH y se correlaciona la expansión de trinucleótidos CAG, la edad de inicio y la presentación clínica.
Población y métodos
Características del Estudio y criterios de selección
Se trata de un estudio analítico de corte transversal realizado entre Agosto 2003 y Junio 2004 por los autores en colaboración con la Unidad de Movimientos Involuntarios y la Unidad de Neurogenética del Instituto Nacional de Ciencias Neurológicas del Perú·.
Población de estudio: 30 pacientes correspondientes a 12 familias provenientes de cinco distritos del Valle de Cañete: Nuevo Imperial, Imperial, Pacarán, Lunahuana y Quilmaná (Figura 1). Las doce familias comprendieron un total de 1397 individuos.
| Figura 1. Los dos primeros mapas muestran el Depatamento de Lima y la provincia de Cañete, el tercer mapa muestra el área de estudio. |
Se incluyeron en el estudio pacientes con confirmación genética por RCP (Reacción en Cadena de Polimerasa) con cuantificación del número de tripletes CAG y que tuviesen información clínica detallada en relación a edad, sexo, historia familiar, hallazgos clínicos y curso de la enfermedad y que aceptaron participar voluntariamente, previo consentimiento informado.
No se incluyeron en el estudio sujetos con otras enfermedad neurodegenerativas o que hubiesen consumido neurolépticos o levodopa por lo menos 6 meses antes; niños o adolescentes con antecedente de faringitis estreptocócica o la negativa del paciente a participar en el estudio.
Protocolo de recolección de datos del estudio
El método de recolección fue la encuesta. A los pacientes que aceptaron voluntariamente participar en el estudio se les entrevistó y se aplicó una encuesta estructurada, precodificada, destinada a la obtención de respuestas sobre el problema en estudio y características de los investigados. Posterior a la encuesta, los pacientes fueron examinados clínicamente en sus domicilios por un mismo examinador (Dr. L.T.R.), se complemento con el Test de Folstein y la Escala Modificada de la EH (UHDRS) (27). Se estableció el diagnóstico de acuerdo a los criterios clínicos propuestos por Folstein en 1986 (28), confirmándose posteriormente por estudios de genética molecular.
Estudios de Genética Molecular
Previa a la extracción de muestras de sangre se obtuvo un consentimiento informado, donde se detalló el uso ulterior de dicha muestra. Se obtuvo una muestra de 5 ml de sangre periférica de los individuos afectados, y de sus familiares en primer grado, extrayéndose ADN mediante los procedimientos habituales. Posteriormente se realizó RCP para determinar el número de repeticiones del triplete CAG de los individuos estudiados.
La RCP específica para los tripletes CAG, se realizó en un volumen de reacción de 25 mL conteniendo 100 ng de ADN genómico, 0,5 mM de cada cebador, 10 mM de Tris (pH 8,3), 5 mM KCl, 1,5 mM MgCl2, 200 mM de dCTP, dTTP y dATP, 80 mM de dGTP y 120m M de 7-deasa-GTP, 10% dimetil sulfóxido y 0,75 U de AmpliTaq Gold. Las reacciones se sometieron a las siguientes condiciones en un termociclador automático: 94°10í (94°1í, 66°1í, 72°1í) x 35 ciclos, 72° 10í. Los cebadores utilizados fueron los siguientes:
“A” HD-F:5í-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3í.
“B” HD-R:5í-AAACTCACGGTCGGTGCAGCGGCTCCTCAG-3í.
Las muestras se procesaron posteriormente en un gel de secuencia de acrilamida al 8% (20:1 acrilamida:bisacrilamida) y 7 M de úrea, el cual fue corrido en una cámara de electroforésis para proteínas Protean II a 300 voltios constantes durante 3 horas y luego fue teñido con nitrato de plata fijándose el gel durante 10 minutos en etanol 10% (v/v) y ácido acético 0.5% (v/v); tinción en solución de nitrato de plata (AgNO3 0.1% w/v) durante 25 minutos con agitación. Luego se realizó un lavado durante tres (03) minutos con agua desionizada y finalmente se reveló con una solución que contiene NaOH 3% (w/v) y 0.1% de formaldehido hasta la identificación de las bandas de los alelos. El revelado se suspende dejando el gel en solución fijadora.
El número de repeticiones CAG se determinó mediante el corrido en paralelo de otros alelos a los que previamente se les determinó el número de repeticiones mediante secuenciamiento.
El procesamiento de la muestra y determinación del número de tripletes se realizó en el Laboratorio de Neurogenética del Instituto Nacional de Ciencias Neurológicas.
Estadística
Los datos fueron procesados en el paquete estadístico Epi Info 6.0. Se usó distribución de frecuencias, porcentajes, promedios, desviación estándar, para la determinación de la magnitud y características de la problemática en estudio. Prueba estadística t de Student para muestras no pareadas para observar si había diferencia significativa en la edad de inicio de la enfermedad en relación al patrón de herencia ya sea paterno o materno. Se consideró como valor estadísticamente significativo p < 0.05 con un intervalo de confidencia de 95%. Para objetivar el nivel de correlación entre variables se utilizó el test de Pearson.
Aspectos Éticos
La información obtenida: historia clínica, fotografías y videos procedió sólo de aquellos sujetos que dieron su consentimiento escrito tras la exposición de los objetivos del estudio.
La información recogida es estrictamente confidencial y de uso exclusivo del grupo investigador.
El resultado del estudio genético fue otorgado al paciente sólo cuando éste lo solicitó y en caso de menores de edad se les otorgará cuando cumplan la mayoría de edad. Los resultados se expresaron de manera estratificada o global sin identificación de personas e instituciones.
Resultados
Resultados Clínicos: De los 30 pacientes, todos de raza mestiza, 17 (56.7%) eran varones y 13 (43.3%) mujeres, entre los 23 y 71 años de edad, con una media de 43.2 años (Tabla 1).
| TABLA 1 |
| EDAD DE INICIO DE LOS SÍNTOMAS EN PACIENTES CON DIAGNÓSTICO DE EH POR GRUPO ETÁREO Y SEXO |
|
| |
Grupo Etáreo
(años)
|
Sexo |
Total de casos
N (%)
|
| |
|
Hombres
N (%) |
Mujeres
N(%) |
|
| |
<20
20-29
30-39
40-49
50-59
60-69
|
1 (3.3)
7 (23.3)
2 (6.7)
3 (10)
2 (6.7)
2 (6.7)
|
-
2 (6.7)
6 (20)
2 (6.7)
1 (3.3)
2 (6.7)
|
1
9 (30)
8 (26.7)
5 (16.7)
3 (10)
4 (13.3)
|
| |
Total |
17(56.7) |
13(43.3) |
30(100) |
|
* Los porcentajes en paréntesis se calculan en función del total de casos.
|
La media de la edad de inicio de los síntomas fue 38.5 años (DS ± 14.32); por sexos la media de edad en varones fue de 36.82 años (DS ± 15.72) y en mujeres de 40.69 años (DS ± 12.62), no existiendo diferencia significativa con respecto a la edad de inicio y sexo (p = 0.473, IC: 95%). Respecto a la edad de inicio de la enfermedad, 1 (3.3%) paciente presentó EH juvenil, 22 (73.4%) presentaron EH del adulto, y 7 (23.3%) correspondieron a EH de inicio tardío.
En relación con el patrón de transmisión, 13 (43.3 %) pacientes refirieron transmisión paterna de la enfermedad, mientras que 17 (56.7%) informaron transmisión materna, no se encontró transmisión de novo. Se observó un inicio más precoz de la enfermedad en aquellos pacientes que tenían un patrón de transmisión paterna (34 años, DS ± 14.7), en comparación de aquellos que tenían transmisión materna (41 años, DS ± 13.7), aunque las diferencias no fueron significativas (p = 0.228, IC: 95%). En el grupo de pacientes con transmisión paterna de la enfermedad, no hubo diferencias significativas (p = 0.129, IC: 95%) en lo que respecta a sexos, teniendo una edad de inicio los varones de 29.88 años (DS ± 15.15) y en mujeres de 42.8 años (DS ± 11.05). El promedio de edad de inicio de la enfermedad para los pacientes con transmisión materna fue para los varones de 43 (DS ± 14.23) años y para las mujeres de 39.38 (DS ± 13.92) años, no se observó diferencias significativas de inicio de la enfermedad con respecto a sexo (p = 0.604, IC: 95%).
La manifestación clínica al inicio de la enfermedad fue el movimiento coreico, el cual se presentó en 30 pacientes (100%), varios de ellos tuvieron alteraciones del comportamiento previas o simultáneas a la corea. Los trastornos psiquiátricos fueron el primer síntoma en 9 pacientes (30%), de los cuales 8 padecieron de irritabilidad y 1 de pérdida de motivación. Ninguno de los pacientes, incluso los de origen tardío de EH, reportó demencia como síntoma de inicio. Un paciente (3.3%) presentó EH juvenil, aunque no comenzó de manera atípica, sino por movimientos involuntarios en miembros superiores y por irritabilidad. Este caso presentó una transmisión paterna de la enfermedad. dieciseis pacientes (53.3%) refirieron trastornos motores (problemas para sostener objetos, torpeza al caminar y dificultad para hablar) como síntomas de inicio. Sólo 1 paciente (3.3%) comunicó pérdida de la memoria como síntoma inicial.
Resultados Moleculares
Se valoró el tamaño de los amplificados (RCP) con los cebadores que flanquean los tripletes CAG. Así se encontró que en los sujetos de nuestro estudio el alelo normal tenía un tamaño del triplete CAG entre 14 y 33 repeticiones con una media de 17.57. Los alelos más frecuentes fueron el 15, 16, 17 y 18 (66.8% de pacientes). Todos los pacientes fueron heterocigotos, presentando 20 de ellos (66.6%) expansiones dentro del límite patológico, el cual osciló entre las 40 y 56 repeticiones con una media de 44.35 y una DS ± 5.25. En 10 (33.3%) pacientes se pudo determinar el número de expansiones dentro del rango conocido como “zona gris o incierta”, en la cual hay un riesgo incrementado de desarrollar la enfermedad. Las expansiones variaron entre 36 y 39 repeticiones con una media de 37.6 y una DS ± 1.43.
En dos ocasiones se pudo estudiar dos generaciones con transmisión paterna. En el primer caso, el progenitor tuvo 39 repeticiones, y dos de sus hijos presentaron un incremento en el número de tripletes presentando 40 y 54 repeticiones respectivamente, observándose fenómeno de anticipación ya que el progenitor inició la enfermedad a los 44 años y los hijos la iniciaron a los 29 y 20 años, con un inicio más temprano en el hijo con mayor número de repeticiones.
En el segundo caso, el progenitor tuvo 39 repeticiones, y sus dos hijos presentaron un número incrementado de repeticiones, 43 en ambos casos, con un inicio precoz de la enfermedad, 24 y 23 años, observándose nuevamente fenómeno de anticipación.
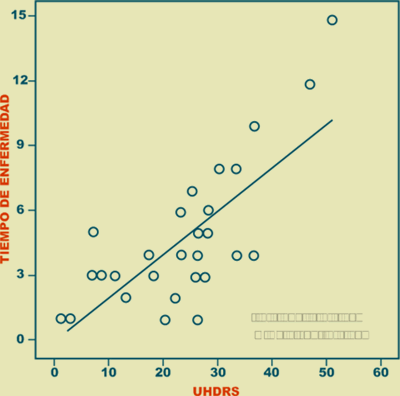
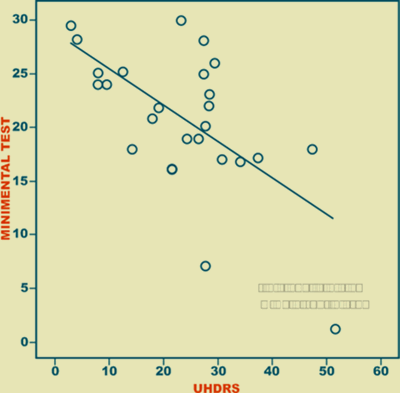
El tiempo de enfermedad presentó una correlación positiva (r = 0.773, p < 0.001) con la severidad de la enfermedad valorada por la escala de UHDRS (Figura 2), mientras que tuvo una correlación negativa (r = -0.678, p < 0.01) con el Minimental Test de Folstein (Figura 3).
Figura 2. Correlación entre el tiempo de enfermedad y la escala UHDRS.
|
Figura 3. Correlación entre el Test de Folstein y la escala UHDRS.
|
Correlación Clínico-molecular
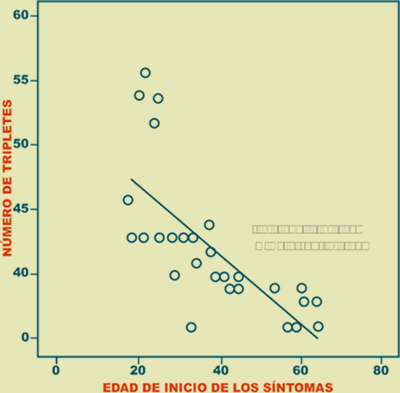
Se observó una correlación inversa entre el número de tripletes CAG y la edad de inicio de la enfermedad (r = - 0.718; p < 0.001) (Figura 4).
El único caso con EH juvenil tenía 46 repeticiones e inició la enfermedad a los 18 años.
Los cuatro casos con mayor expansión (52, 54 y 56 repeticiones) iniciaron su enfermedad en la tercera década de la vida.
De los 10 pacientes con tripletes dentro del rango conocido como “zona incierta”, 7 presentaron inicio tardío de la EH, con edades de inicio entre los 53 y 64 años.
Se obtuvo información sobre la edad de inicio de los progenitores en 17 de los 30 pacientes, observándose que en aquellos pacientes con transmisión paterna de la enfermedad hubo una tendencia a adelantarse la edad de inicio, con una diferencia de medias de 6.55 años (34.5 años: DS ± 14.7 frente a 41.4 años: DS ± 7.24).
No hubo diferencias en el tamaño medio de la expansión en función del patrón de transmisión, siendo para la transmisión paterna 42 (DS ± 4.69), y para la transmisión materna 42.18 (DS ± 6.03).
Figura 4. Correlación entre el número de tripletes CAG y la edad de inicio de la enfermedad.
|
Discusión
En 1993, Gusella et al. descubrieron el gen causante de la EH mediante la clonación del gen HD; estudio que permite un diagnóstico seguro de esta enfermedad, mediante la determinación del número de tripletes CAG del gen de la huntingtina (16).
El presente trabajo constituye el primer estudio de genética molecular realizado en nuestro país en pacientes con EH. La mayoría de los pacientes presentaron la forma de inicio clásica de la enfermedad en la cuarta década de la vida (24, 29-31). Foroud et al.(32) en un estudio que incluyó a 2068 pacientes reportó edad de inicio a los 40 años, con un rango entre los 2 a 75 años. Adams et al. (33) en 1988 estudiaron 611 casos y la edad promedio de inicio fue de 38.7. Folstein (34) en 1987 informa un promedio de edad de inicio de 40.3 años. Myers (14) en 1985 en 243 pacientes observó promedio de edad de inicio de 40.9 años.
Más importante que la edad de inicio de la enfermedad son los extremos del inicio de esta. En nuestro estudio, se presentaron casos con manifestaciones juveniles y tardías, en proporción del 3.3 y 23.3% respectivamente, lo cual es comparable a lo reportado por Walker (35) y otros recientes estudios (32, 33, 36).
El tamaño de los alelos expandidos oscilaron entre las 40 y 56 repeticiones con una media de 44.35. El alelo normal de mayor tamaño tenía 33 repeticiones y el menor 14. A primera vista estos datos sugieren que la determinación del tamaño del triplete repetitivo supondría el diagnóstico totalmente seguro de la enfermedad. Sin embargo, tal y como se demuestra en otros estudios, entre las distribuciones normal y patológica existe una franja de “reducida penetrancia” o “zona incierta” (37). Según estudios realizados en individuos sanos y afectados, parece claro que un tamaño del triplete CAG menor o igual a 35 repeticiones se puede considerar normal, mientras que por encima de las 40 repeticiones no se han encontrado casos de individuos con larga supervivencia y sanos (38). En nuestro estudio 10 pacientes con diagnóstico clínico de EH, tenían alelos en el rango de las 36 y 39 repeticiones, presentando todos inicio tardío de la enfermedad. Estos resultados difieren de lo reportado por Lima (39) et al. en cuyo estudio no encontró pacientes con características clínicas de EH y que tuvieran alelos en el rango conocido como “zona incierta”.
En cuatro de nuestros pacientes se pudo obtener el número de tripletes de sus padres, observándose un obvio incremento en el número de tripletes. Sin embargo, un incremento en la expansión, no siempre se asocia con términos de anticipación de la edad de inicio. Andrew (40) et al. identificaron 8 pacientes en los cuales la diferencia de las edades de inicio con sus padres fue de 10 años o más; sin embargo, las diferencias en la expansión de nucleótidos entre padres e hijos fue 2 o menos. Todo esto sugeriría que en pacientes con un inicio de la enfermedad en la adultez otros factores aparte del número de tripletes pueden jugar un rol importante en la determinación de la edad de inicio de la enfermedad (41).
En nuestro estudio se encontró que había una correlación negativa entre el Test de Folstein y el UHDRS, mientras que existía una correlación positiva entre este último y el tiempo de enfermedad, esto concuerda con el estudio realizado por el Huntington Study Group (27), quienes encontraron que esta escala se correlacionaba cuando se comparaba con pruebas de desempeño cognoscitivo; además a mayor tiempo de enfermedad se obtenían mayores valores en la escala UHDRS, lo cual concuerda con nuestro estudio. Illarioshkin et al. (42) encontraron una correlación significativa entre el número de tripletes CAG y la tasa de progresión de síntomas tanto neurológicos como psiquiátricos. Esto difiere de lo reportado por Claes (43) et al. quienes no encontraron una correlación significativa entre el número de tripletes y el modo de progresión de la enfermedad.
Hemos encontrado una correlación significativa entre la edad de inicio y el número de repeticiones del triplete CAG, al igual que en otras series (36, 40). En nuestro estudio, el único paciente con EH juvenil tuvo 46 repeticiones, mientras que otros pacientes con mayor número de repeticiones si bien no iniciaron la enfermedad antes de los 20 años, manifestaron síntomas en la tercera década de la vida.
Se ha reportado diferencias en la estabilidad del número de tripletes CAG de acuerdo al patrón de transmisión de la enfermedad, siendo más inestable la transmisión paterna con tendencia a un incremento en el número de tripletes de una generación a otra (44, 45). Esta tendencia no ha sido observada en nuestro trabajo, en el cual no hubo diferencias significativas entre el número de tripletes de los pacientes con transmisión paterna (CAGn 42) y los de transmisión materna (CAGn 42.18).
Conclusiones
Aunque el número de repeticiones esta fuertemente correlacionado con la edad de inicio, estos datos no serán particularmente útiles en el diagnóstico presintomático excepto en aquellos pacientes con un alto número de repeticiones. Si el incremento del número de tripletes es evidente en un paciente, esto significará que la persona en riesgo ha heredado la mutación asociada con la EH, pero no nos proveerá ninguna información concerniente al estado clínico actual del paciente.
Agradecimientos
Agradecemos la colaboración prestada por la Ingeniero Químico María Marca Isabel y el Biólogo Olimpo Ortega Dávila del Laboratorio de Neurogenética del Instituto Nacional de Ciencias Neurológicas, sin la ayuda de ellos no hubiera sido posible la realización de este trabajo.
Referencias bibliográficas
- Harper P. Huntington's Disease. Major Problems in Neurology. ed 2da, Londres, Saunders, 1991.
- Martin J. Huntington's Disease: New approaches to an old problem. Neurology 1984;34:1059-1072.
- Snowden JS, Craufurd D, Griffiths HL. et al. Awareness of Involuntary Movements in Huntington Disease. Arch Neurol 1998;55:801-805.
- Jankovic J, Ashizawa T. Tourettism Associated with Huntington's Disease. Mov Disord 1995;10:103-105.
- Tian JR, Zee DS, Lasker AG. et al. Saccades in Huntington's disease: Predictive tracking and interaction between release of fixation and initiation of saccades. Neurology 1991;41:875-881.
- Deus-Yela J, Pujol J, Espert R. Neuropsychological deterioration in Huntington's disease. Rev Neurol 1997;25:1257-1268.
- Paulsen JS, Ready RE, Hamilton JM. et al. Neuropsychiatric aspects of Huntington's disease. J Neurol Neurosurg Psychiatry 2001;71:310-314.
- Anderson KE, Louis ED, Stern Y. et al. Cognitive correlates of obsessive and compulsive symptoms in Huntington's disease. Am J Psychiatry 2001;158:799-801.
- Redondo -Vergé L. Deterioro cognitivo en la enfermedad de Huntington. Rev Neurol 2001;32:82-85.
- Jason GW, Suchowersky O, Pajurkova EM. et al. Cognitive Manifestations of Huntington's Disease in relation to Genetic Structure and Clinical Onset. Arch Neurol 1997;54:1081-1088.
- Gutekunst CA, Norflus F, Hersch S. Recent advances in Huntington's disease. Current Opinion in Neurology 2000;13:445-450
- Pericak -Vance MA, Elston RC, Conneally PM. Age of onset heterogeneity in Huntington's disease families. Am J Med Genet 1983;14:49-59.
- Feigin A, Kieburtz K, Bordwell K. et al. Functional Decline in Huntington's Disease. Mov Disord 1995;10:211-214.
- Myers RH, Sax DS, Schoenfeld M. et al. Late onset of Huntington's disease. J Neurol Neurosurg Psychiatry 1985;48:530-534.
- Mohmood S, Sherwani A, Khan F. et al. DNA trinucleotide repeat expansion in neuropsychiatric patients. Medical Science Monitor 2003;9:237-245.
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993;72:971-983.
- Leroi I, O'Hearn E, Marsh L. et al. Psychopathology in patients with degenerative cerebellar diseases: a comparison to Huntington's disease. Am J Psychiatry 2002;159:1306-1314.
- Metzler M, Helgason CD, Dragatsis I. et al. Huntingtin is required for normal hematopoiesis. Hum Mol Genet 2000;12:387-394.
- Bates G. Huntingtin aggregation and toxicity in HuntingtonEs disease. Lancet 2003;361:1642-1644.
- Everett CM, Wood NW. Trinucleotide repeats and neurodegenerative disease. Brain 2004;127:2385-2405.
- Petersen A, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington's disease. Exp Neurol 1999;157:1-18.
- Laccone F, Engel U, Holinski-Feder E. et al. DNA analysis of Huntington's disease: Five years of experience in Germany, Austria and Switzerland. Neurology 1999;53:801-806.
- Saavedra A. Sobre un caso de Corea de Huntington. Rev. Neuropsiquiatría 1950;2:232-239.
- Cuba JM, Castro C, Benzaquen M. Sobre la Epidemiología de la Corea de Huntington en el Perú. Rev. Neuropsiquiatría 1983;46:114-120.
- Cuba JM. A focus of Huntington's chorea in Perú. Rev Neurol (Paris) 1986;142:151-153.
- Cuba JM, Torres L. Huit Arbres Généalogiques de Chorée de Huntington au Peróu. Rev Neurol (Paris) 1989;145:482-484.
- Grup HS. Unified Huntington's Disease Rating Scale: realiability and Consistency. Mov Disord 1996;11:136-142.
- Folstein SE, Leigh RJ, Parhad IM. et al. The diagnosis of Huntington's disease. Neurology 1986;36:1279-1283.
- Vonsattel JP, DiFiglia M. Huntington Disease. J Neuropathol Exp Neurol 1998;57:369-384.
- Martin J, Gusella J. Huntington's disease: Pathogenesis and Management. N Engl J Med 1986;315:1267-1276.
- Burguera JA, Solis P, Salazar A. Estimate of the prevalence of Huntington disease in the Valencia region using the capture-recapture method. Rev Neurol 1997;25:1845-1847.
- Foroud T, Gray J, Ivashina J. et al. Differences in duration of Huntington's disease based on age at onset. J Neurol Neurosurg Psychiatry 1999;66:52-56.
- Adams P, Falek A, Arnold J. Huntington's Disease in Georgia: age at onset. Am J Hum Genet 1988;43:695-704.
- Folstein SE, Chase GA, Wahl WE. at el. Huntington disease in Maryland: clinical aspects of racial variation. American Journal of Human Genetics 1987;41:168-179.
- Walker DA, Harper PS, Wells CE. et al. Huntington's Chorea in South Wales. A genetic and epidemiological study. Clin Genet 1981;19:213-221.
- Solís M, Palau F, Burguera JA. Enfermedad de Huntington: estudio clínico y genético en una población española. Neurologia 1995;10:362-366.
- Brinkmann RR, Mezei MM, Thielmann J. et al. The likehood of being affected with Huntington disease by a particular age for a specific CAG size. Am J Hum Genet 1997;60:202-210.
- Hernando I, Álvarez V, García Martínez, A. et al. Huntington chorea: clinical and molecular analysis in Asturian patients. Neurologia 1999;14:11-15.
- Lima e Silva TC, Serra HG, Bertuzzo CS. et al. Molecular Diagnosis of Hungtington Disease in Brazilian Patients. Arq Neuropsiquiatr 2000;58:11-17.
- Andrew SE, Goldberg YP, Kremer B. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nature Genetics 1993;4:398-403.
- Wexler NS, Lorimer J; Porter, J. et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A 2004;101:3498-3503.
- Illarioshkin SN, Igarashi S, Onodera O. et al. Trinucleotide repeat length and rate of progression of Huntington's disease. Ann Neurol 1994;36:630-635.
- Claes S, Van Zand K, Legius E. et al. Correlations between triplet repeat expansion and clinical features in Huntington's disease. Arch Neurol 1995;52:749-753.
- De Rooij KE, De Koning Gans PA, Skraastad MI. et al. Dynamic mutation in Dutch Huntington's disease patients: increased paternal repeat instability extending to within the normal size range. J Med Genet 1993;30:996-1002.
- Trottier Y, Biancalana V, Mandel JL. Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J Med Genet 1994;31:377-382.
1 Jefe del Departamento de Enfermedades Neuro-degenerativas y de la Unidad de Movimientos Involuntarios del Instituto
Nacional de Ciencias Neurológicas. 2 Médicos Rotantes del Instituto Nacional de Ciencias Neurológicas.
3 Jefa de Unidad de Neurogenética del Instituto Nacional de Ciencias Neurológicas. |