Hígado graso :
Nuevos conceptos en patogénesis y manejo clínico
Matín Tagle(1)
Introducción
La esteatohepatitis no alcohólica (EHNA) es un estadío intermedio en el espectro de la enfermedad hepática grasa no alcohólica (EHGNA), una enfermedad hepática crónica que histológicamente recuerda a la hepatitis alcohólica, pero que ocurre en individuos que no son bebedores. Es la causa más frecuente de elevación inespecífica de transaminasas en la práctica diaria, y la evidencia que la señala como etiología de la cirrosis “criptogénica” es cada vez mayor. Como en la esteatosis alcohólica, la EHGNA varía desde la esteatosis en el extremo más benigno del espectro hasta la cirrosis en el extremo opuesto, donde la mayoría de morbilidad y mortalidad asociada a enfermedad hepática ocurre (1) . En este artículo revisaremos los avances en la patofisiología y el manejo de esta importante entidad.
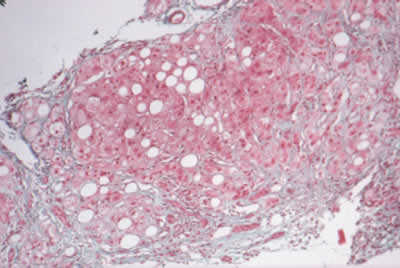
 |
| Figura 1. Esteatohepatitis no alcohólica (RHNA). Obsérvese el severo infiltrado graso y las bandas anchas de tejido fibrótico e infiltrado inflamatorio. (Cortesía: Dra Yolanda Scavino, Clínica Anglo Americana, Lima). |
Espectro de Enfermedad e Historia Natural
En el HGNA la grasa se acumula dentro de los hepatocitos. Se considera que tiene un curso benigno siempre y cuando no se asocie a cambios en la arquitectura hepática, necrosis y fibrosis. La cirrosis resulta cuando la fibrosis altera la arquitectura hepática normal, lo cual ocurre en algunos pacientes. Tal como ocurre en otras enfermedades hepáticas, este avance de hígado graso “inflamatorio” a fibrosis y cirrosis toma probablemente décadas. Aún se desconoce por qué algunos individuos desarrollan cambios más agresivos que otros, y esto es materia de investigación (2).
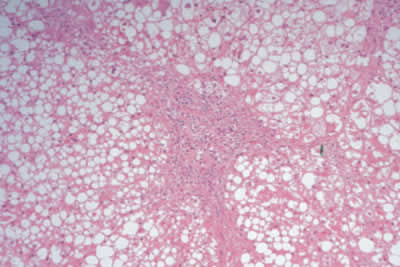
| Figura 2. Esteatohepatitis no alcohólica (RHNA). La tinción tricrómica fibrosis moderada a severa y esteatosis macrovesicular. (Cortesía: Dra Yolanda Scavino, Clínica Anglo Americana, Lima). |
Patogénesis
Rol central de la resistencia a la Insulina y la complejidad de los mecanismos implicados
La EHGNA está fuertemente asociada a obesidad (3). La obesidad es un componente común del síndrome de insulinoresistencia (también llamado síndrome metabólico ó Síndrome X). La obesidad y el síndrome metabólico están asociados entre sí porque el tejido graso es un productor importante de factores que regulan el metabolismo y la remodelación de tejidos. Tales factores incluyen a algunas citoquinas (factor de necrosis tumoral alfa f TNF alfa -, adiponectina, y al inhibidor del activador del plasminógeno f PAI-1, asi como mediadores neuroendocrinos (leptina, resistina, norepinefrina, angiotensina). Los transtornos que ocurren en el síndrome metabólico son el resultado de una producción anormal de citoquinas que regulan las respuestas inflamatorias. Los sujetos con el síndrome metabólico generalmente tienen un exceso de citoquinas proinflamatorias con respecto a los factores antiinflamatorios (4, 5).
La obesidad es un transtorno común del síndrome metabólico y el exceso de adiposidad intraabdominal principalmente contribuye al daño tisular debido a su rol en la regulación de la respuesta inflamatoria (6). Por lo menos tres de estos factores: ácidos grasos, adinopectina y factor de necrosis tumoral Alpha (TNF-Alpha) modulan la respuesta inflamatoria hepática. La adiponectina interactúa con su receptor en el hepatocito para inhibir la entrada de ácidos grasos, al mismo tiempo que estimula la oxidación de ácidos grasos y su exportación (7). Estas acciones tienden a inhibir como resultado final la acumulación de grasa en el hepatocito. También tiende a promover la sensibilidad a insulina. El TNF alpha es una citoquina pro-apoptótica que cumple un rol clave en el reclutamiento de células inflamatorias al tejido injuriado, al mismo tiempo que promueve la insulino-resistencia. Interesantemente, la adiponectina y el TNF-alpha son mutuamente antagónicos, al inhibir la producción y actividad uno del otro (8).
Estudios en animales han probado que la producción desbalanceada de citoquinas derivadas de la grasa, tales como TNF alpha y adiponectina, promueven daño en muchos tejidos, incluyendo al hígado. Xu y col. (9). han demostrado una alta relación TNF:adiponectina en ratones ob/ob , un modelo genético de obesidad, diabetes y EHNA. Estos ratones tiene deficiencia de leptina, una hormona reguladora de la saciedad, produciéndose una obesidad mórbida en dichos animales. Tratando a ratones ob/ob con adiponectina mejora su hiperglicemia e insulino- resistencia, reduciendo la hepatomegalia, esteatosis hepática, niveles de ALT y TNF alpha. En estudios paralelos, ellos demostraron que la terapia con adiponectina tambien disminuye TNF alpha y mejora la esteatohepatitis inducida por alcohol. Posteriormente, Masaki y col. (10) demostraron similares beneficios del tratamiento con adiponectina en ratones KK-Ay, otro modelo genético de obesidad que tiene aumento de TNF alpha y predisposición a enfermedad hepática. La esteatosis resultante del disbalance adiponectina f TNF alpha y la insulino-resistencia promueve la oxidación intracelular de ácidos grasos, que a su vez generan especies reactivas en oxígeno (también conocidos como radicales libres ). Esto genera un estado de stress oxidativo que puede dar lugar a la muerte del hepatocito.
Dado que la reducción de la actividad de TNF alpha fue una consecuencia consistente del tratamiento con adiponectina en todos estos estudios, no sorprende que tratando a los ratones ob/ob con anticuerpos para neutralizar el TNF alpha también mejore su EHNA (11). Feldstein y col. (12) recientemente reportaron que los ratones que son genéticamente deficientes en receptores tipo 1 de TNF están protegidos contra EHNA a pesar de desarrollar obesidad, insulino-resistencia e hiperleptinemia cuando consumen dietas altas en sucrosa. Por lo tanto, la relación TNF: adiponectina es alta en distintos modelos animales de EHNA, y ésta mejora con tratamientos que aumenten la adiponectina o que reduzcan la actividad de TNF.
Estudios en humanos con EHGNA y EHNA también demuestran que un disbalance entre adiponectina y TNF alpha tienen un rol en la patogénesis de EHNA. Una comparación entre pacientes con EHGNA y EHNA (13) mostró que la probabilidad de tener EHNA aumentaba 6 veces por cada 5 microgramo/ml de disminución en niveles de adiponectina. En ese mismo estudio, el riesgo de EHNA se duplicaba por cada picogramo/ml de aumento en TNF-alpha.
Otro hallazgo importante en la patogénesis de la EHGNA es el incremento en la actividad hepática del factor nuclear kappa-beta (NF-kB) y su activador inhibidor kappa kinasa beta (IKK-B), que funciona como un sistema regulador proinflamatorio, supraregulando la transcripción de una gran variedad de mediadores de la inflamación. La inhibición del NF-kB atenúa la expresión genética de citoquinas y la activación de las células de Küpffer en respuesta a dietas altas en grasas (14). Fedelstein demostró que los ácidos grasos pueden activar directamente la vía IKK-B/NF-kB en hepatocitos vía un mecanismo dependiente de lisosomas (13). La desestabilizacion de los lisosomas libera una cisteína proteasa, la Catepsina B en el citosol. Esto a su vez lleva a la activación del NF f kB vía IKK-B y por lo tanto a un incremento en el TNF-alpha.
Otro potencial vínculo entre esteatosis, activación del factor NF-kB, injuria hepática y resistencia a la insulina en EHGNA ha sido propuesto por un estudio reciente que demuestra que el stress en el retículo endoplásmico es una característica de los ratones ob/ob (15). El retículo endoplásmico es una red membranosa que provee un ambiente especial en donde las proteínas secretorias logran una estructura tridimensional correcta. Cuando las demandas metabólicas aumentan, también lo hace la demanda del retículo endoplásmico. El stress biológico como la hiperinsulinemia, hipoxia y exceso de ácidos grasos puede alterar la homeostasis del retículo endoplásmico y desencadenar la llamada “respuesta del retículo endoplásmico al stress”, que resulta en la activación de una serie de citoquinas y disfunción mitocondrial.
Las bacterias intestinales también pueden modular la actividad de TNF-alpha y adiponectina. Estudios con ratones sin gérmenes intestinales y sus contrapartes normales sugieren que la flora intestinal regula los genes intestinales y hepáticos que promueven la absorción de lípidos así como la síntesis hepática de ácidos grasos (16).
Resumiendo, hay muchas razones por las cuales se desarrolla síndrome metabólico durante la obesidad. La grasa visceral provee TNF y ácidos grasos libres directamente al hígado. Esto inhibe la actividad de la adiponectina promoviendo EHGNA e insulino-resistencia. El hígado graso tiene pobre capacidad para aclarar el lipopolisacárido derivado de las bacterias intestinales, permitiendo que esta sustancia con gran potencial generador de citoquinas entre al hígado. El tejido graso periférico sobre-expuesto a lipopolisacárido estimula más liberación de TNF-alpha y ácidos grasos inhibiendo la libreación de adiponectina, generando insulino-resistencia.
Con respecto a la fibrogénesis, las células esteladas o de Ito, se encuentran íntimamente ligadas a reparación tisular hepática bajo condiciones de injuria. Estas células expresan receptores en su superficie para: leptina, adiponectina, agonistas adrenérgicos y angiotensina (17, 18). Cuando estas células se activan son capaces de producir una gran matriz colágena condicionando fibrosis. La norepinefrina aumenta el número de células esteladas en el modelo de ratón obeso deficiente en leptina (ob/ob), así como de la citoquina fibrogenética, el factor de crecimiento y transformación (TGF-beta). La leptina no sólo es un inductor endógeno de norepinefrina (20), sino también está regulada por la angiotensina (21) . El tratamiento de ratones ob/ob con leptina corrige su deficiencia de leptina y normaliza sus respuestas fibrogénicas a hepatotoxinas. Interesantemente, debido a que el suplemento de ratones ob/ob con leptina corrige la obesidad y elimina la EHNA, existe la posibilidad de que la propensión a inflamación hepática y fibrogénesis sean dos fenómenos independientes. Se ha demostrado que el tratamiento de ratones ob/ob con norepinefrina induce la expresión hepática de las citoquinas profibrogénicas TGF-beta y aumenta la expresión de genes de producción de colágeno en este modelo animal (22).
Manejo clínico
El primer paso en diagnosticar EHGNA en personas con enzimas hepáticas elevadas es descartar otras causas, tales como hepatitis viral, autoimmune o enfermedades hereditarias. La tabla 1 muestra las causas principales de elevación de aminotransferasas y las pruebas de descarte correspondientes. El paciente debe ser evaluado para las características del sindrome metabólico, y establecer una sospecha clínica de hígado graso. Como es sabido, ningun exámen de imágenes puede distinguir EHGNA de EHNA. En la ecografía, exámen accesible fácilmente por su relativo bajo costo y su difundido uso, puede verse hiperrefringencia del paránquima hepático. El HGNA tambien puede sospecharse cuando hay hepatomegalia. Se recomienda una biopsia hepática si el diagnóstico de fibrosis avanzada tiene implicancias pronósticas para nuestro paciente. Por ejemplo, el hallazgo de cirrosis hepática nos llevaría a indicar una endoscopía alta para descartar várices esofágicas y al monitoreo semestral con Alfa Feto Proteína y Ecografía hepática para supervigilancia de carcinoma hepatocelular
| TABLA 1 |
| ENTIDAD |
ESTUDIO DE LABORATORIO |
|
Hepatis B
Hepatitis C
|
HBsAg, HBc Ab
Anti-HCV
|
Hepatitis autoinmune
|
ANA, Anti-músculo Liso, globulinas
|
Hemocromatosis Hereditaria
|
Hierro, TIBC, Ferritina, estudios genéticos |
| Medicamentos hepatotóxicos |
Historia Clínica |
| Enfermedad Celíaca |
Anti-Transglutaminasa Tisular
|
Enfermedad de Wilson
|
Ceruloplasmina |
| Deficiencia de Alfa-1 Antitripsina |
Niveles séricos, genotipo
|
| Alcohol |
AST/ALT. Historia Clínica |
|
.
Dado que la mayoría de pacientes con EHGNA son portadores del síndrome metabólico, el manejo óptimo de éste incidirá positivamente sobre la esteatosis. Algunos paneles de expertos internacionales indican que el ejercicio regular es esencial para logar una pérdida de peso duradera (22). Idealmente el paciente debe hacer ejercicio de intensidad moderada por 60 minutos tres veces por semana. Sin embargo, un plan que comienza con 20 minutos de caminata diaria es recomendable. La ingesta calórica debe ser individualizada para lograr un déficit de 500 a mil calorías diarias (23). La ingesta de grasa saturada debe reducirse, y el total de grasa ingerida debe disminuir a menos del 30% de la ingesta calórica total. Asimismo, debe disminuirse la ingesta de azúcares refinadas y aumentar la de fibra soluble. Hay evidencia que señala que la pérdida de peso disminuye los niveles de leptina, Interleukina 6 y TNF alpha en diabéticos (24). La ingesta de alcohol debe ser minimizada en estos pacientes para reducir el riesgo de hepatopatía. A este repecto, Ruhl y col. (25) demostraron en un estudio de más de 13 mil personas en los Estados Unidos, que los obesos (indice de masa corporal > 30 kg/m2 ) que bebían más de dos tragos al día tenían un riesgo de 5.4 veces mayor de tener elevación de aminotransferasas comparado con los abstemios que tenían índice de masa corporal de < 25 kg/m2. La reducción eficaz de peso con medidas dietéticas y ejercicio solamente logra mejorar no solamente los valores de aminotransferasas, sino tambien la histología de los pacientes con esteatohepatitis no alcohólica. Así, Huang y col. (26) estudiaron 23 pacientes con EHNA demostrada histológicamente, y los sometieron a 12 meses de dieta estricta y ejercicio regular. Quince de lo pacientes fueron re-biopsiados, demostrándose clara mejoría del score de NASH en 9 de ellos. Medicamentos como el Orlistat (27) han demostrado eficacia en mejorar los cambios histológicos de EHNA, y aparentemente este efecto se debe no sólo a la reduccion de peso per se sino también a la reducción en la absorción de ácidos grasos intestinales hacia la circulación portal, mejorando así la sensibilidad a la insulina (28).
Hay dos clases de medicamentos utilizados para el manejo de la insulino-resistencia: Las biguanidas (Metformina) y las Tiazolidinedionas como la Rosiglitazona y la Pioglitazona. En un estudio, la Metformina demostró mejoría de los niveles de aminotransferasas en pacientes con diagnóstico presuntivo de esteatosis hepática, sin embargo no se realizó biopsias hepáticas (29). Las Tiazolidinedionas (TZDs) actúan mediante los receptores PPAR y mejoran la sensibilidad a la insulina. Neuschwander-Tetri y col. (30) estudiaron 30 pacientes con EHNA y los trataron con Rosiglitazona 4 mg biD por 48 semanas. Se logró una mejoría del score NASH en 45% de los pacientes, sin embargo el 67% de los ellos aumentó de peso, siendo éste el efecto secundario más frecuente. En otro estudio de diseño similar con Pioglitazona (31) se obtuvo mejoría histológica en dos terceras partes de los pacientes, pero el aumento de peso también fue frecuente.
Algunos medicamentos que presumiblemente confieren protección al hepatocito han sido estudiados últimamente. Betaine es un precursor de la S-adenosil metionina (SAME), un hepatoprotector. En un estudio 10 pacientes recibieron betaine por 12 meses en dos dosis diarias. Siete de los 10 completaron tratamiento, notándose mejoría en niveles de aminotransferasas y de histología hepática (32). El ácido Ursodeoxicólico (UDCA) es un ácido biliar hidrofílico de uso conocido en enfermedades colestásicas. Sin embargo su rol en EHGNA ha sido cuestionado por un estudio randomizado, placebo controlado a 2 años donde no hubo diferencias con placebo en cuanto a respuesta bioquímica e histológica (33). Actualmente se espera los resultados de otro estudio en curso con UDCA a dosis altas, así como de otros estudios con antioxidantes (vitamina C y vitamina E) conducidos por el Instituto Nacional de Salud de los Estados Unidos (NIH).
La cirugía bariátrica es a veces medicamente necesaria porque es el único método probado para lograr el control de peso a largo plazo en el obeso mórbido (IMC > 40 kg/m2 ). Muchas series han documentado que luego de cirugía bariátrica los pacientes pierden peso rápido y lo mantienen por más de 24 meses, perdiendo el 50% del exceso de peso en los primeros 6 meses y el 77% del mismo en un año, con mejoría sustancial de complicaciones asociadas como hipertensión, apnea de sueño, y displipidemia (34, 35). Es de esperarse que la cirugía bariátrica tenga un efecto benéfico en la esteatosis hepática que los obesos mórbidos tienen universalmente. Mathurin y col. (36) reportan 121 pacientes evaluados 1 año luego de perder un promedio de 27 kg de peso mediante gastroplastía (“banda gástrica”) ó bypass gastro-yeyunal. Hubo directa relación entre insulino-resistencia y esteatosis severa. Se reportó mejoría en el score de esteatosis, y la EHNA desapareció en 75% de los pacientes que tuvieron dicho diagnóstico antes de la cirugía. Curiosamente sin embargo, el score de fibrosis empeoró ligeramente luego de un año de la cirugía. Si bien es cierto es claro el beneficio de la cirugía bariátrica en lo que respecta a insulino-resistencia y la salud en general, no se sabe si estos beneficios se traducen en mejoría de la morbilidad y mortalidad hepática. Se necesita estudios de seguimiento a largo plazo para esclarecer esta duda.
Conclusiones
La esteatosis hepática es una causa importante de morbilidad a nivel mundial, y se asocia con frecuencia a resistencia a la Insulina. Últimamente se han esclarecido algunos mecanismos por medio de los cuales la grasa visceral juega un papel predominante en la patogénesis de la resistencia a la insulina y la generación de citoquinas pro-inflamatorias. Hay evidencia que algunos pacientes con hígado graso no alcohólico pueden progresar a cirrosis, por eso la importancia de investigar su presencia en cualquier paciente con sobrepeso u obesidad o elevación persistente de aminotransferasas (mal llamada antiguamente “transaminitis”, término que resta importancia a la investigación que debe hacerse en todo paciente con elevación mínima de aminotransferasas). No hay un tratamiento establecido para el hígado graso no alcohólico, pero la piedra angular del manejo de esta entidad es la optimización del peso corporal y la corrección del síndrome metabólico cuando está presente. El rol de los agentes farmacológicos es lamentablemente muy limitado hasta el momento, sucediendo lo mismo con la cirugía bariátrica pese a sus beneficios en otros aspectos metabólicos. Se necesita más estudios para desarrollar una terapia efectiva de esta enfermedad cuya incidencia está indudablemente en ascenso.
Bibliografía
- Brunt EM. Nonalcoholic Steatohepatitis. Semin Liv Dis 2004;24:3-20.
- McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:521-533.
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S. et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003;37:917-992.
- Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003;144:3765-3773.
- Atzmon G, Yang XM. et al. Differential gene expression between visceral and subcutaneous fat depots. Horm Metab Res; 2002;34:622.
- Garg R, Tripathy D, Dandona P. Insulin resistance as a proinflammatory state: mechanisms, mediators, and therapeutic interventions. Curr Drug Targets 2003;4:487-492.
- Stefan N, Stumvoll M. Adiponectin--its role in metabolism and beyond. Horm Metab Res 2002;34:469-474.
- Bruun JM, Lihn AS, Verdich C, Pedersen SB, Toubro S, Astrup A, Richelsen B. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab 2003;285:E527-533.
- Xu A, Wang Y, Keshaw H. et al. The fat-derived hormona adiponectin alleviated alcoholic and nonalcoholic fatty liver disease in mice. J Clin Invest 2003;112:191.
- Masaki T, Chiba S. et al. Adiponectin protects LPS-induced liver injury through modulation of TNF alpha in KK-AY obese mice. Hepatology 2004;40:117.
- Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song X, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve non-alcoholic fatty liver disease. Hepatology 2003;37:343-350.
- Feldstein AE, Wernerburg NW. et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF alpha expression via a lysosomal pathway. Hepatology 2004;40:185.
- Hui M, Hodge A. et al. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004; 40:46.
- Samuel VT, Liu Z-X, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem, 2004;279:32345-32353.
- Ozcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action and Type 2 diabetes. Science 2004;306:457-461.
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718-15723.
- Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct 2003;28:105-112.
- Bataller R, Gines P, Nicolas JM, Gorbig MN, Garcia-Ramallo E, Gasull X, Bosch J. et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000;118:1261-1265.
- Oben JA, Roskams T, Yang S, Lin H, Sinelli N, Li Z, Torbenson M, Thomas SA, Diehl AM. Norepinephrine induces hepatic fibrogenesis in leptin deficient ob/ob mice. Biochemical Biophysical Research Communications 2003;308:284-292.
- Commins SP, Marsh DJ, Thomas SA, Watson PM, Pedgett MA, Palmiter R, Gettys TW. Norepinephrine is required for leptin effects on gene expression in brown and white adipose tissue. Endocrinology 1999;140:4772-4778.
- Correia ML, Haynes WG. Leptin, obesity and cardiovascular disease. Curr Opin Nephrol Hypertens 2004;13:215-223.
- Executive summary of the clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults. Arch Intern Med. 1998;158:1855-1867.
- Yancy WS, Jr., Olsen MK, Guyton JR, Bakst RP, Westman EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Ann Intern Med. 2004;140:769-777.
- Monzillo LU, Hamdy O, Horton ES. et al. Effect of lifestyle modification on adipokine levels in obese subjects with insulin resistance. Obes Res 2003;11(9):1048-1054.
- Ruhl CE, Everhart JE. Joint effects of body weight and alcohol on elevated serum Alanine Aminotransferase in the United States population. Clin Gastroenterol Hepatol 2005;3:1260-1268.
- Huang MA, Greenson JK, Chao C. et al. One year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: A pilot study. Am J Gastroenterol 2005;100:1072-1081.
- Harrison SA, Ramrakhiani S, Brunt EM, Anbari MA, Cortese C, Bacon BR. Orlistat in the treatment of NASH: a case series. Am J Gastroenterol. 2003;98:926-930.
- Zelber-Sagi S, Kessler A, Brazowsky E. et al. A Double-blind, randomized, placebo-controlled trial of Orlistat for the treatment of Non Alcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol 2006;4:639-644.
- Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet 2001; 358:893-894.
- Neuschwanter-Tetri BA, Brunt EM, Wehmeier KR. et al. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand Rosiglitazone. Hepatology 2003;38:1008-17.
- Promrat K, Lutchman G, Uwaifo GI. et al. A pilot study of Pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004;39:188-196.
- Abdelmalek MF, Angulo P, Jorgensen RA, Sylvestre PB, Lindor KD. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: results of a pilot study. Am J Gastroenterol.2001;96:2711-2717.
- Lindor KD, Kowdley K, Heathcote E. et al. Ursodeoxycholic acid for the treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology 2004;39:770-778.
- Buchwald H, Avidor Y, Braunwald E. et al. Bariatric surgery: a systematic review and meta-analysis. JAMA 2004;292:1724-1737.
- Maggard MA, Shugarman LR, Suttorp M. et al. Meta-analysis: surgical treatment of obesity. Ann Intern Med 2005;142:547-559.
- Mathurin P, González F, Kerdraon O. et al. The evolution of severe steatosis alter bariatric surgery is related to insulin resistance. Gastroenterology 2006;130:1617-1624.
1 Profesor Asociado de Medicina de la Universidad Peruana Cayetano Heredia (UPCH). |