Características clínicas de la atrofia multisistémica: Un análisis de 17 casos.
Luis Torres Ramírez (1), Erick Alberto Guevara Silva (2), Carlos Cosentino Esquerre (3)
Resumen
Introducción:
La Atrofia Multisistémica (AMS) es una enfermedad neurodegenerativa caracterizada por parkinsonismo (AMS-P), disfunción cerebelosa (AMS-C) e insuficiencia autonómica en varias combinaciones. El objetivo de este estudio fue describir las características clínicas de esta enfermedad en un centro de neurológico de referencia. Población y métodos: Estudio descriptivo, longitudinal y retrospectivo de pacientes con AMS atendidos desde 1998 al 2008, según la base de datos del Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas. Resultados: Diecisiete pacientes cumplieron los criterios diagnósticos. La edad promedio de inicio de enfermedad fue 58.5 años. Encontramos un predominio del sexo masculino (M:F = 2.4:1) y del subtipo AMS-P (76.4%). Los hallazgos iniciales más frecuentes fueron los signos parkinsonianos o cerebelosos. Los estudios de neuroimagen se realizaron en 12 pacientes, de los cuales 10 mostraron alteraciones. La mayoría de los pacientes presentaron síntomas autonómicos (58.8%), pero solo un paciente fue sometido a un test de función autonómica, encontrándose ortostatismo. En 3 pacientes se realizaron EMG de esfínter anal, mostraron denervación del núcleo de Onuf. Conclusiones: Se encontró un predominio del sexo masculino y de la forma AMS-P que concuerdan con la literatura. Nuestra investigación se basó estrictamente en los últimos criterios establecidos; sin embargo, puede no haberse incluido algunos pacientes con síntomas iniciales y sin diagnóstico preciso; en ningún caso se tuvo confirmación neuropatológica. A pesar de estas limitaciones, es la primera vez que se describen las características clínicas de pacientes con AMS en un centro neurológico de referencia en Perú.
Palabras clave: Atrofia multisistémica, atrofia olivopontocerebelosa, degeneración estrionígrica, disfunción autonómica,
Abstract
Introduction: Multiple System Atrophy (MSA) is a neurodegenerative disease characterized by a combination of parkinsonism (MSA-P), cerebellar dysfunction (MSA-C), and autonomic failure. Our study aimed to describe the clinical features of multiple system atrophy (MSA) in a neurological referral setting. Population and methods: A descriptive, longitudinal and retrospective study was designed, which included every patient diagnosed as AMS since 1998 at 2008 from Neurodegenerative Disease Department database of the Instituto Nacional de Ciencias Neurológicas. Results: Seventeen patients fulfilled the diagnostic criteria. The mean age at onset of the disease was 58.5 years. We found a predominance of males (M:F = 2.4:1) as well as AMS-P cases (76.4%). The most common initial presenting features were parkinsonism or cerebellar signs. Neuroimaging was done in 12 patients, 10 of which were abnormal. Most of patients documented autonomic symptoms (58.8%); however, autonomic function testing was performed only in one patient in whom orthostatism was recorded. Anal sphincter electromyogram was done in 3 patients, all of them indicated denervation of Onuf´s nucleus in the sacral cord. Conclusions: A predominance of male as well as MSA-P was found, these findings coincide with literature data. As our study strictly adhered to the last consensus criteria. However, missed cases can occur as a result of the early cases misdiagnosed as other types of parkinsonism; also, not all our patients had MRI of the brain performed and no neuropathological confirmation was available. Despite these limitations, this is the first review of the clinical characteristics of MSA patients in a neurological referral setting in Peru.
Key words: Autonomic dysfunction, multiple system atrophy, olivopontocerebellar atrophy, parkinsonism, striatonigral degeneration.
Introducción
La Atrofia Multisistémica (AMS) es una enfermedad neurodegenerativa que ha sido descrita durante décadas con diferentes nombres de acuerdo a las características clínicas predominantes; así, la atrofia olivopontocerebelosa (OPCA), por tener primordialmente manifestaciones cerebelosas, fue descrita por primera vez por Dejerine y Thomas en el año 1900. La forma con predominio de disautonomía fue reportada por Shy y Drager en 1960; finalmente la que cursa con características parkinsonianas, degeneración estrioní-grica (SND), fue comunicada por van der Eecken en 1960 y por Adams en 1964 (1-4). Pero fue en el año 1969 cuando se introduce el término AMS, para denominar a los cuadros neurodegenerativos progresivos que ocurren esporádicamen- te y se caracterizan por parkinsonismo, disfunción cerebelosa e insuficiencia autonómica en diferentes proporciones (5). El cuadro parkinsoniano incluye a la bradiquinesia con rigidez, inestabilidad postural, temblor, pobre respuesta a levodopa o buena respuesta pero solo al inicio. La disfunción cerebelosa consiste en ataxia de la marcha y de extremidades, compro- miso del habla y de los movimientos extraoculares. La insuficiencia autonómica se manifiesta como hipotensión ortostática, disfunción eréctil, constipación y disminución de la sudoración; así también, incontinencia urinaria y nicturia (6). Sin embargo, actualmente se simplifica en dos subtipos clínicos: AMS-P y AMS-C de acuerdo a si el trastorno motor es predominantemente parkinsoniano o cerebeloso (7). Este trastorno afecta a ambos sexos, inicia frecuentemente en la edad adulta y tiene un promedio de supervivencia de 9 años desde la aparición del primer síntoma (8).
De etiología incierta se caracteriza a la histopatología por la presencia de inclusiones citoplasmáticas gliales (ICG) (9), y hace algunos años se descubrió mediante técnicas de inmunotinción a la α-sinucleína como marcador patológico de esta entidad por lo que actualmente se la clasifica como una alfasinucleopatía junto con la enfermedad de Parkinson, demencia por cuerpos de Lewy y otros (10).
Según estudios epidemiológicos la prevalencia es de 1.94-4.5 por 100000 habitantes y con una incidencia promedio de 3 nuevos casos por 100 000/año (11-13). Han sido propuestos muchos criterios diagnósticos pero recién en 1998 se acordó por consenso las guías diagnósticas (Tabla 1 y 2)(7).
Estas características clínicas que fueron establecidas utilizando los consensos actuales sobre el diagnóstico de AMS no han sido evaluadas en Sudamérica, particularmente no existen publicaciones sobre esta enfermedad en nuestro país, constituido por una población étnica y culturalmente diversa, es por ello que nosotros analizamos el espectro clínico y las características de la AMS en pacientes que acudieron a nuestra institución.
Estas características clínicas que fueron establecidas utilizando los consensos actuales sobre el diagnóstico de AMS no han sido evaluadas en Sudamérica, particularmente no existen publicaciones sobre esta enfermedad en nuestro país, constituido por una población étnica y culturalmente diversa, es por ello que nosotros analizamos el espectro clínico y las características de la AMS en pacientes que acudieron a nuestra institución.
Material y método.
Este estudio descriptivo de corte longitudinal y retrospectivo incluyó a todos los pacientes identificados y tratados como AMS desde 1998 al 2008 en el Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas. Se revisaron las historias clínicas de cada paciente para obtener las características clínicas, demográficas y resultados de exámenes auxiliares los cuales fueron anotados en una ficha estructurada y precodificada. La categoría diagnóstica se determinó de acuerdo a los criterios diagnósticos publicados en 1999 (Tablas 1 y 2).
El estudio incluyó a todo paciente con diagnóstico de AMS, hecho por dos neurólogos con experiencia en enfermedades neurodegenerativas y en base a los criterios diagnósticos mencionados arriba.
No se incluyeron aquellas historias clínicas con datos o estudios incompletos.
Análisis estadístico
Se utilizó el programa estadístico SPSS 15.0. Los resultados son expresados de manera estratificada o global sin identificación de personas.
Resultados
Revisamos los registros médicos de 17 pacientes con diagnóstico de AMS en base a los criterios de consenso, todos fueron diagnosticados y tratados por neurólogos con expe- riencia en enfermedades neurodegenerativas. Del total de pacientes, 10 cumplieron los criterios para AMS probable y 7 se catalogaron como AMS posible.
El promedio de edad de inicio de los síntomas fue de 58.5 años (rango, 36 a 79 años; DS=11.3) y al momento del diagnóstico, 62.1 años (rango, 39 a 80 años; DS=11.1).
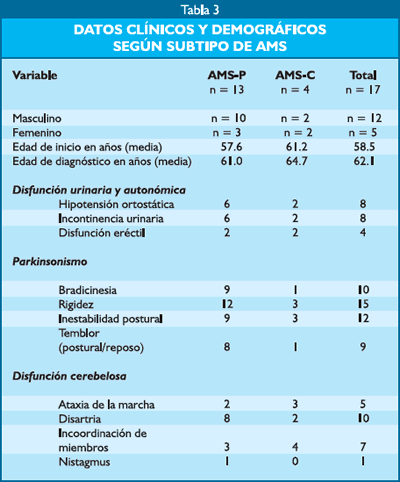
Las principales características clínicas y demográficas se describen en la tabla 3, la mayoría de los pacientes fueron varones, así mismo el 76.4% pertenecieron al tipo AMS-P.
Cuatro pacientes no presentaron ninguna comorbilidad, del resto (76.4%) las condiciones comórbidas más comunes fueron: HTA (4), ictus isquémico (2), hipertrofia prostática (8), distonía cervical primaria (1), depresión (1), bronquitis crónica (1), asma (1) y fibrilación auricular (1).
Los síntomas de inicio de la enfermedad fueron solo parkinsonianos o cerebelosos en un 64.7% (10 pacientes parkinsonianos y 1 cerebeloso); enfermedad con compromiso de más de un sistema y 2 pacientes debutaron solo con síntomas autonómicos (Figura 1). Si se toma en cuenta la última vez que el paciente fue evaluado en el Instituto, el 100% de pacientes ya tenían síntomas parkinsonianos, el 58.8% (10 pacientes) síntomas autonómicos, 47% (8 pacientes) se objetivaban síntomas cerebelosos y el 35.2% (6 pacientes) ya habían desarrollado piramidalismo.
Seis pacientes no regresaron a sus controles luego de recibir el diagnóstico de AMS, los 11 restantes fueron seguidos en un tiempo que varió de 1 a 4 años con un promedio de 2.5 años, en todos ellos el cuadro fue progresivo, dos pacientes fallecieron durante el internamiento en fases avanzadas de la enfermedad, en ningún caso se realizó estudio anatomopatológico.
Catorce (82.3%) pacientes recibieron levodopa, de los cuales 11 pertenecieron al subtipo AMS-P, llegando a una dosis máxima de 1000mg/día. Algún grado de mejoría subjetiva al inicio de la enfermedad fue reportado en 9 pacientes del subtipo AMS-P y en 2 pacientes del subtipo AMS-C. Otros fármacos utilizados fueron: selegilina, pergolida, ácido valproico, pramipexol y entacapone, sin eficacia considerable.
 |
| Traducido de “Consensus statement on the diagnosis of multiple system atrophy” (8). |
 |
Traducido de “Consensus statement on the diagnosis of multiple system
atrophy” (8). |
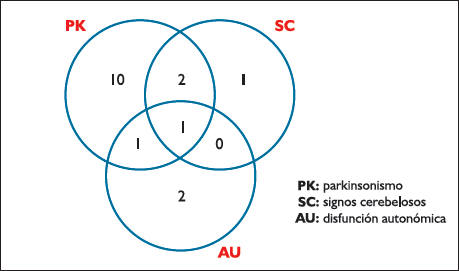 |
Figura 1. Frecuencia de varias combinaciones de las características clínicas
iniciales. |
Las manifestaciones de disfunción autonómica en algún momento de la enfermedad se presentaron en 10 pacientes (58.8%): constipación en 2 y disminución de la sudoración en 2. Solo en un caso se realizó la prueba de la mesa basculante donde se evidenció hipotensión ortostática.
Los signos piramidales se hallaron en 6 pacientes (35.2%). Un paciente presentó nistagmus y dos pacientes mostraron lentitud de los movimientos oculares verticales. Dos pacientes manifestaron alteración del sueño (insomnio con excesivo sueño en el día) por hipo persistente o sensación de atragantarse con la saliva.
La electromiografía del esfínter anal se realizó en 3 pacientes y en todos ellos los hallazgos fueron sugestivos de denervación del núcleo de Onuf.
Los estudios por neuroimágenes fueron realizados en 12 pacientes (70.5%), de los cuales 5 fueron por resonancia magnética (RM) y 7, por tomografía cerebral (TC). De los pacientes que se hicieron RM, una fue normal y en las otras se informaron: incipiente compromiso trófico de corteza cerebelosa (mujer, AMS-C), atrofia cortical cerebelosa simétrica con hiposeñal de ambos globus pallidum y putamen (varón, AMS-C), leve atrofia olivopontocere- belosa (mujer, AMS-P) y atrofia córtico-subcortical a predominio temporal bilateral (varón, AMS-P). Los hallazgos por TC fueron: leve atrofia cortical, nódulos calcificados y otras lesiones inespecíficas, un paciente mostró TC normal.
Discusión
Encontramos 17 pacientes que cumplieron con los criterios clínicos probables (10 casos) o posible (7 casos) para AMS. Puede haber dificultades en el diagnóstico de AMS en parte porque 10 a 20% de los casos se presentan como un parkinsonismo relativamente puro incluso con respuesta a levodopa (14). Mientras que el uso de los criterios actuales para AMS puede mejorar la precisión del diagnóstico en la primera visita, en la última entrevista estos criterios no son más precisos que el diagnóstico final del neurólogo (15). La sensibilidad reportada para estos criterios de consenso utilizando exámenes neuropatológicos se en- cuentra en un rango de 63% a 88%.
Nuestro estudio mostró un evidente predominio masculino (M:F, 2.4:1), tal como se ha visto en otros estudios (8,16). También encontramos un predominio del subtipo AMS-P (76.4%), lo cual concuerda con estudios previos (8) y con un metanálisis de estudios realizados principalmente en poblaciones norteamericanas y europeas (17); no obstante, investigaciones en países como Singapur (16), Japón (18) y Alemania (19) mostraron un predominio del subtipo AMS-C.
La edad promedio de inicio de la enfermedad (58.5 años) es ligeramente mayor al encontrado en el trabajo de Wenning et al. (8) pero menor al encontrado por Jamora et al. (16). La menor edad hallada fue de 36 años.
Encontramos un mayor número de pacientes puramente parkinsonianos que cerebelosos al inicio de la enfermedad, mientras que en otro estudio asiático se encontró estos síndromes en la misma proporción, pero en un trabajo de 100 pacientes en Inglaterra, 48 tuvieron síntomas parkinsonianos de inicio y solo 9 fueron cerebelosos (8,16). En términos de características autonómicas encontramos igual número de pacientes con disturbios urinarios y con hipotensión ortostática, mientras que en otros estudios los transtornos urinarios fueron mayores (20,21); solo 4 de nuestros pacientes mostraron disfunción eréctil, a diferencia del trabajo de Wenning et al. quienes tuvieron una población con casi la totalidad de varones con este problema (8).
Los tests de función autonómica y neuroimágenes no son parte de los criterios diagnósticos pero frecuentemente son utilizados para sustentar el diagnóstico y excluir otras condiciones. La mayoría de nuestros pacientes tuvieron un estudio de imágenes, ya sea tomografía o resonancia, solo en un caso se encontró atrofia cerebelosa y anomalías del putamen. Un anillo hiperintenso en el borde lateral del putamen ha sido reportado en alrededor de un tercio de pacientes con AMS en el estudio de Wenning et al. (8). Sin embargo, la resonancia magnética puede ser normal hasta en el 20% de los casos (22). Los tests de función autonómica pueden ayudar a corroborar los signos y síntomas del paciente o a detectarlos cuando son asintomáticos mas no puede diferenciar entre AMS y otras enfermedades que también cursan con transtornos autonómicos como la enfermedad de Parkinson (23). En nuestro Instituto se cuenta con la mesa basculante a partir desde el año 2006 y desde entonces se ha utilizado en varios pacientes con otros síndromes pero de nuestros pacientes solo uno pudo utilizarla confirmando la sospecha de hipotensión ortotática. La electromiografía del esfínter anal indicó denervación de núcleo de Onuf en 42 de 46 pacientes estudiados con esta técnica en un estudio (8) y en 44 de 52 pacientes en otro (24), nosotros encontramos que los únicos tres pacientes que se hicieron dicho estudio mostraron esta anormalidad. Por otro lado, en un trabajo retrospectivo de Paviour et al; se encontró que de 30 pacientes con AMS a quienes se le realizó este examen solo uno fue normal, por ello sugieren que una EMG normal puede ser utilizado como un argumento en contra de este diagnóstico, pero los hallazgos anormales deben ser tomados con cautela ya también puede presentarse en la enfermedad de Parkinson y Degeneración Corticobasal (25). Este examen podría ser de valor en pacientes en quienes se sospecha de AMS pero no presentan hipotensión ortostática. Finalmente, hasta que no tengamos varios estudios prospectivos bien diseñados en relación a este examen auxiliar, su utilidad clínica continuará siendo debatida.
Existen además otras características clínicas como disfonía, estridor laríngeo y distonía que presentaron algunos de nuestros pacientes pero que es más frecuente si se revisa la literatura, así lo demuestra una investigación donde la disartria ocurrió en la mayoría de los pacientes, alteración de los movimientos sacádicos (68%), estridor respiratorio (34%), antecollis (15%) y mioclonías (31%) (8).
Es posible que durante la revisión no se hayan incluido muchos pacientes que aún están en estudio sin un diagnóstico definido o que son considerados como enfermedad de Parkinson u otro tipo de parkinsonismo y que posteriormente la evolución nos confirme que se trata de AMS-P como sucedió inicialmente con varios de nuestros pacientes, siempre existirán casos en quienes sea difícil diferenciar entre estas dos patologías sin embargo un número de investigadores han sugerido ciertas características clínicas que pueden ayudarnos, como el inicio simétrico de los síntomas, la ausencia de temblor y la falta de respuesta a levodopa y la presencia de estridor laríngeo que apuntan más hacia una AMS (8, 26).
Este trabajo tiene las limitaciones propias de un estudio retrospectivo. Ningún paciente tuvo confirmación hispatológica. El seguimiento fue limitado en algunos pacientes y los exámenes auxiliares así como el tratamiento no fueron aplicados de forma estandarizada. Además el pequeño número de pacientes de una sola institución no permita establecer la verdadera prevalencia de esta enfermedad. Una evaluación prospectiva y multicéntrica sería ideal para definir mejor la historia natural, respuesta al tratamiento y pronóstico clínico de los pacientes con AMS en Perú, así como su incidencia y prevalencia.
A pesar de estas limitaciones, esta es la primera descripción en nuestro país de un grupo de pacientes con AMS, evaluados en un centro neurológico de referencia, utilizando los criterios de consenso. Nuestro estudio reveló un predominio de pacientes con ASM-P en contraste con otras publicaciones; y un predominio masculino y edad de inicio similar a otras publicaciones.
Bibliografía
- Dejerine J, Thomas A. L'atrophie olivopontocerebelleuse. Nouv Iconogr Salpet 1900;13:330-370
- Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathological study. Arch Neurol 1960;2:511-527.
- van der Eecken H, Adams RD, van Bogaert L. Striopallidal-nigral degeneration. A hitherto undescribed lesion in paralysis agitans. J Neuropathol Exp Neurol 1960;19:159-161.
- Adams RD, van Bogaert L, van der Eecken H. Striato-nigral degeneration. J Neuropathol Exp Neurol 1964;23:584-608.
- Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 1969;32:28-34.
- Robertson D, Shannon J, Jordan J, et al. Multiple system atrophy: new developments in pathophysiology and therapy. Parkinsonism Relat Disord 2001;7:257- 260.
- Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94-98.
- Wenning GK, Ben-Shlomo Y, Magalaes M, et al. Clinical features and natural history of multiple system atrophy: an analysis of 100 cases. Brain 1994;117:835-845.
- Lantos PL. Multiple system atrophy. Brain Pathol 1997;7:1293-1297.
- Tu PH, Galvin JE, Baba M, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol 1998;44:415-422.
- Bower JH, Maraganore DM, McDonnell SK, et al. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 1997;49:1284-1288.
- Chrysostome V, Tison F, Yekhlef F, et al. Epidemiology of multiple system atrophy: a prevalence and pilot risk factor study in Aquitaine, France. Neuroepidemiol 2004;23:201-208.
- Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 1999;354:1771-1775.
- Hughes AJ, Daniel SE, Ben-Shlomo Y, et al. The accuracy of diagnosis of parkinsonian síndromes in a specialist movement disorder service. Brain 2002;125:861-870.
- Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002; 59:1486-1491.
- Jamora RDG, Gupta A, Tan AKY, et al. Clinical characteristics of patients with multiple system atrophy in Singapore. Ann Acad Med Singapore 2005;34:553-537.
- Ben-Shlomo Y, Wenning GK, Tison F, et al. Survival of patients with pathologically proven multiple system atrophy: A meta-analysis. Neurology 1997;48:384-393.
- Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125:1070-1083.
- Schulz JB, Klockgether T, Petersen D, et al. Multiple system atrophy: natural history, MRI morphology, and dopamine receptor imaging with 123IBZM-SPECT. J Neurol Neurosurg Psychiatry 1994;57:1047-1056.
- Kaufmann H. Multiple system atrophy. Curr Opin Neurol 1998;11:351-355.
- Sakakibara R, Hattori T, Uchiyama T, et al. Urinary dysfunction and orthostatic hypotension in multiple system atrophy: which is the more common and earlier manifestation? J Neurol Neurosurg Psychiatry 2000;68:65-69.
- Sandroni P, Ahlskog J, Fealey R, et al. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991;1:147-155.
- Riley D, Chelimsky T. Autonomic nervous system testing may not distinguish multiple system atrophy from Parkinson´s disease. J Neurol Neurosurg Psychiatry 2003;74:56-60.
- Wang H. Relationship between the autonomic dysfunction of multiple system atrophy and external anal sphincter electromyography. Clinical Neurophysiology 2006;117:S121-S336.
- Paviour D, Williams D, Fowler C, et al. Is Sphincter Electromyography a Helpful Investigation in the Diagnosis of Multiple System Atrophy? A Retrospective Study With Pathological Diagnosis. Mov Disord 2005;20:1425-1430.
- Hughes A, Daniel S, Ben-Shlomo Y, et al. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement.
1 Jefe del Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Médico Neurólogo. Universidad Nacional Mayor de San Marcos (UNMSM). Lima, Perú.
3 Jefe de la Unidad de Movimientos
Involuntarios del Instituto Nacional de Ciencias Neurológicas. Lima, Perú. |