|
|
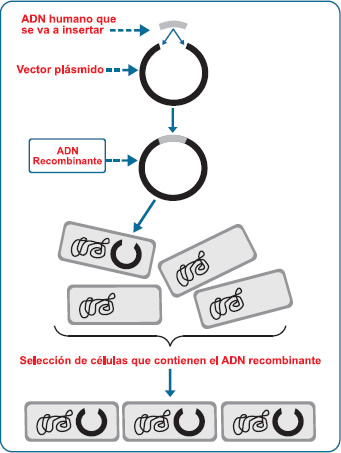
Medicamentos Biologicos Fernando De Mora (1),Rosa Torres(2) ResumenLos biológicos, es decir, medicamentos obtenidos de organismos vivos, no son nuevos. La historia proporciona varios ejemplos de extractos animales o humanos que se utilizan para prevenir o tratar enfermedades humanas. En consecuencia, los médicos han estado conscientes, por siglos, del valor terapéutico de nuestras propias moléculas. La dificultad radicó, muchas veces, en cómo obtener estos compuestos propios o similares a los propios. La biotecnología, una tecnología mediante la cual organismos vivos manipulados se utilizan para generar productos útiles tales como fármacos, proporcionó una respuesta revolucionaria. Sabemos cómo crear genéticamente por ingeniería bacterias, levaduras, células de insectos o mamíferos para sintetizar moléculas humanas, las llamadas proteínas terapéuticas recombinantes humanas. Los anticuerpos monoclonales murinos y humanizados contra antígenos humanos también son productos biotecnológicos. El número de fármacos biotecnológicos que se están comercializando, y aquéllos que se utilizan en ensayos clínicos o que están a la espera de autorización, está creciendo de manera exponencial. Actualmente, aún estamos en los inicios de una nueva era en farmacoterapia, cuyo final es imposible de ver. Los farmacólogos deben seguir el ritmo de estos cambios y desarrollar nuevas habilidades. Probablemente, incluso tengan que desafiar antiguas suposiciones, con la finalidad de investigar nuevas moléculas. Utilizando un enfoque fácil y entendible, este artículo de revisión vuelve a tratar bio-conceptos y da énfasis a la dimensión real del desafío. Palabras clave: Biomedicamentos; biológicos; productos biofarmacéuticos; biosimilares; ADN recombinante; proteínas terapéuticas. ¿Qué son los biológicos?La búsqueda de nuevos fármacos ha llevado a los seres humanos a explorar los bosques tropicales y el lecho marino, y diseñar nuevas moléculas, de manera más o menos exitosa, pero con frecuencia bastante a ciegas. Sin embargo, en el caso de varias de las enfermedades o síndromes seleccionados como objetivo, probablemente no hayamos tenido que hacer un esfuerzo de tal magnitud para descubrir fármacos activos, ya que la solución estaba adecuadamente dentro de nuestro alcance, en el interior de nuestro propio cuerpo, en forma de moléculas con una función conocida que se puede convertir en medicamentos. La idea no es nueva. En realidad, la historia reciente proporciona varios casos. Por ejemplo, desde los años 40, se sabe que los glucocorticoides que nosotros generamos, cuando se administran como fármacos, tienen un efecto antiinflamatorio e inmunosupresor, sobre el cual se ha comprobado que es beneficioso en los procesos autoinmunes, alérgicos e inflamatorios, así como en la prevención del rechazo de trasplantes de órganos (1). El misterio que quedaba por resolver, además de identificar un posible beneficio terapéutico para las moléculas endógenas, era cómo obtenerlos y purificarlos. En el caso de los esteroides, aprendimos a producirlos mediante síntesis química y, actualmente, contamos con un amplio rango de agentes que más o menos se asemejan a sus contrapartes naturales. El caso de las hormonas proteicas resultó ser más difícil y, con frecuencia, era necesario dirigirse hacia los tejidos y órganos humanos o animales en los que se originan. Un ensayo bien documentado con la terapia de reemplazo hormonal data del año 1896, cuando una mujer fue tratada eficazmente con extracto de tejido ovárico bovino cargado con estrógenos administrado por vía oral (2,3). Sin embargo, tal vez el ejemplo más impactante del uso terapéutico de tejido animal fue la insulina (4). En el año 1922, insulina extraída de páncreas bovino demostró regular la concentración de la glucosa en la sangre en un paciente diabético que no podía sintetizar la hormona (posteriormente, dada su gran similitud con la hormona humana, la insulina del porcino se convirtió en la fuente principal de insulina hasta finales del siglo pasado). Agentes de este tipo se conocen como biológicos. Si bien, en farmacología, el término biológico se aplicó original y básicamente a los hemoderivados y vacunas, hoy en día se le utiliza como sinónimo de medicamento biológico (o biológico, en realidad) y biomedicamento, para definir, de acuerdo con la Agencia Europea de Medicamentos (EMA) (5), un fármaco o medicamento obtenido de un organismo vivo, que se entiende que comprende desde seres humanos hasta bacterias, e incluso virus. Esto establece una distinción fundamental entre los medicamentos biológicos y los medicamentos utilizados actualmente con mayor frecuencia, es decir, aquellos pequeños productos elaborados mediante síntesis química y ya conocidos como fármacos clásicos, tradicionales o convencionales, cuya producción no involucra organismos o células, ni como intermediarios ni como fuente. Técnicamente, los medicamentos biológicos incluyeron de ese modo agentes conocidos, tales como las gammaglobulinas (6) (?-globulinas), es decir, anticuerpos o inmunoglobulinas aisladas de sangre humana para prevenir o combatir infecciones, antibióticos (7) (cuando se obtuvieron directamente de bacterias u hongos), vacunas elaboradas a partir de extractos de microorganismos atenuados o, más recientemente, heparinas de bajo peso molecular (LMWH) (8) (obtenidas de tejido pulmonar bovino o mucosa gástrica porcina), y factor de coagulación VIII cuando se extrae del plasma humano (9). Por lo tanto, ¿qué es lo novedoso en el tema de los medicamentos biológicos? La novedad, al menos en términos del orden del día para los avances científicos, es que ahora tenemos acceso a técnicas más refinadas y eficientes que permiten una producción más confiable de biológicos especí- ficos en el laboratorio, en cantidad suficiente, tal como lo veremos inmediatamente. Además, el mayor conocimiento de la actividad endógena de algunos de estos productos ha llevado a la industria farmacéutica a realizar una enorme cantidad de proyectos de investigación, desarrollo e/o innovación (R&D&i), ayudando de ese modo a forjar los inicios de una era verdaderamente nueva en la farmacología: la era de los biomedicamentos (o productos biofarmacéuticos; un término acuñado en 1980, aunque con un significado ligeramente diferente). El deseo del psicoterapeuta de conocerse a sí mismo nunca ha sido tan provechoso para la medicina; dicho deseo nos coloca en una posición privilegiada para ampliar las fronteras de la terapia farmacológica. Medicamento biotecnológico: Un tipo de medicamento biológicoUn medicamento biológico implica, de alguna manera, más que ingeniárselas para obtener una molécula endógena en el laboratorio. El amplio grupo de medicamentos biológicos incluye a un subgrupo (fármacos derivados de procesos biotecnológicos) que se está ampliando exponencialmente. Estos fármacos conforman el tema del presente artículo, y preferimos simplificarlos denominándolos productos biotecnológicos. La biotecnología, es decir, la tecnología basada en organismos vivos o moléculas biológicas (enzimas o anticuerpos) que se utilizan o manipulan intencionalmente para obtener productos provechosos (10), ha iniciado una nueva área en la farmacología. Los agentes biotecnológicos se producen, por consiguiente, de organismos vivos, debido a que son biológicos, pero para producir lo anterior, los organismos deben haber sido manipulados de alguna manera. Por ejemplo, el material genético de una entidad como Escherichia coli se puede alterar de tal manera que, sorprendentemente, se pueda producir una proteína humana. Esto se conoce como tecnología de ADN recombinante (rADN), que forma parte de la ingeniería genética, o manipulación genética, que a su vez pertenece al campo más amplio de la biología molecular. A pesar de que siempre existe cierta similitud en su estructura química y composición, los productos obtenidos mediante técnicas biotecnológicas no siempre están planeados para ser copias exactas de moléculas endógenas humanas. En consecuencia, los anticuerpos monoclonales contra autoantígenos producidos en ratones que han sido reproducidos para propósitos de investigación (manipulados) y que han sido tratados anteriormente (manipulados) también son agentes biotecnológicos (11). Estos son dos ejemplos claros de técnicas biotecnológicas que sirven a la farmacología y, por consiguiente, a la medicina. La primera compañía en producir una molécula tera-péutica comercialmente viable usando la biotecnología fue Genentech, fundada por Robert Swanson y Herbert Boyer en el año 1976. Desde entonces, con el lanzamiento de la insulina humana recombinante (abreviada como rHu o rh) (1982), la hormona de crecimiento humano recombinante o rhGH (es decir, somatotropina recombinante) (1985), y la eritropoyetina humana recombinante, también conocida como EPO o epoetina (1989), ha estado en curso una carrera imparable en el campo de los productos biofarmacéuticos. Vale la pena recordar que, por definición, la biotecnología también incluye el proceso de fermentación utilizado para producir alimentos y bebidas, tales como la cerveza, vino y queso; sin embargo, no abordaremos esta aplicación en este documento. De lo mencionado anteriormente, se puede deducir que, debido a que se generan en organismos vivos, todos los fárma-cos que tienen un origen biotecnológico son medicamentos biológicos, pero no todos los medicamentos biológicos son fármacos biotecnológicos. Esta dualidad entre un fármaco biotecnológico y un medicamento biológico se observa claramente en el factor de coagulación VIII. El factor VIII humano, administrado a los hemofílicos para restaurar la hemostasis, se puede obtener, como mínimo, de dos maneras. Una involucra la extracción del plasma humano (9); el fármaco se conoce entonces como un hemoderivado y se considera, por definición, como un medicamento biológico, pero no como un fármaco biotecnológico. Sin embargo, si el mismo factor VIII se produce usando la tecnología del rADN (12), se describe como un fármaco biotecnológico. En realidad, el SIDA, que apareció a inicios de los años 80 (y cuya transmisión se atribuyó a la transfusión de sangre y, por lo tanto, a la terapia que involucra a los hemoderivados), fue uno de los principales impulsos para realizar investigaciones en la producción biotecnológica de proteínas en sangre humana en lugar de aislarlas de la sangre. Sin embargo, a pesar de que se había admitido que habría un cambio con respecto a los procedimientos de extracción de sangre para que los procesos recombinantes generen hemoderivados tales como el factor VIII y otros, unas cuantas compañías aún comercializan exitosamente productos aislados del plasma (13). ADN recombinante: Pasado, presente y futuro No hay duda de que el potencial de la biotecnología en la farmacología es ilimitado. Estamos ahora en el umbral de una era cuyo final es imposible ver, mientras que ya agentes de la segunda y tercera generación están abriéndose paso en el mercado. Hoy en día, a pesar de que los fármacos biotecnológicos no se han integrado socialmente, ni siquiera por varios profesionales de la salud, se está desarrollando una nueva generación de fármacos biotecnológicos, los cuales serán utilizados ampliamente por los estudiantes del presente y futuro. Un área de la biotecnología implica la obtención de pequeños fragmentos de ácidos nucleicos (oligonucleótidos) que, al actuar en el genoma, pueden restringir o reforzar la expresión de genes específicos para componer lo que se ha venido conociendo como terapia génica (14). Esta área también incluye el uso de células madre para fines terapéuticos (el denominado medicamento regenerativo) (15,16). Ya se están llevando a cabo experimentos en los campos de la ciencia básica e investigación clínica, de tal manera que la biotecnología pueda proporcionar incluso más opciones terapéuticas innovadoras. A pesar de que los fármacos biotecnológicos varían ampliamente en términos de su estructura química, la mayoría son proteínas, o al menos moléculas con un componente peptídico considerable. Se espera que estas proteínas terapéuticas dominen el mercado correspondiente a los fármacos biotecnológicos en los próximos años. Los anticuerpos monoclonales son proteínas, independientemente de cómo se producen; sin embargo, el impulso detrás de los avances en los fármacos biotecnológicos es la técnica del rADN, que hace posible elaborar péptidos y proteínas (proteínas recombinan-tes) humanos extremadamente puros para fines terapéuticos, de manera más eficiente. Los avances realizados en este campo, desde los años 70, llevaron a la introducción en 1982 por Eli Lilly de la insulina humana recombinante producida por Genentech. Ésta fue la primera molécula recombinante que se lanzó como un fármaco, y reemplazó a la insulina aislada del páncreas porcino, la más utilizada hasta entonces (17). Las ventajas del rADN incluyen su capacidad de superar muchos de los peligros implicados en la administración de moléculas extraídas de tejidos humanos y no humanos. Se puede observar un ejemplo en el tratamiento de niños con enanismo congénito que, hasta los años 80, se trató con la hormona de crecimiento derivada de hipófisis de cadáveres (somatotropina o somatropina). Esto produjo a veces la enfermedad de Creutzfeldt-Jakob (encefalopatía espongiforme) (18), un inconveniente que se superó con el desarrollo de la somatotropina humana recombinante. La idoneidad terapéutica de las proteínas recombinantes frente a enfermedades severas está fuera de duda. Tomar en cuenta, por ejemplo, las ventajas ofrecidas por la rhEPO en el tratamiento de pacientes anémicos con insuficiencia renal crónica (19), el éxito de filgrastim recom- binante (factor estimulante de la colonia de granulocitos o G-CSF) en pacientes con cáncer que reciben quimioterapia (20) y la utilidad del interferón creado por ingeniería (INF)-a en el tratamiento de la hepatitis (21). También se emplean otras citocinas (por ejemplo, el IFN-ß o interleucina [IL]-2), vacunas biotecnológicas como la reciente vacuna contra el papilomavirus humano, anticuerpos monoclonales que requieren esta tecnología en su proceso de producción, factores de coagulación, enzimas, así como otras proteínas y péptidos (frecuentemente hormonales), tales como la insulina y somatotropina, como se mencionó anteriormente. El mayor interés en estos fármacos por parte de los investigadores y médicos generales significa que también reciben la atención de entidades regulatorias y autoridades sanitarias. Como siempre es el caso en biomedicina, los últimos adelantos en la farmacología biotecnológica son el resultado de décadas, incluso siglos, de esfuerzos por parte de muchos investigadores. Los principales puntos de referencia se tornaron obvios durante el siglo XX, con el otorgamiento de varios Premios Nobel. Al controversial otorgamiento del Premio Nobel en Medicina y Fisiología en 1923 a Banting y McLeod por su descubrimiento de la insulina (22), le siguió el reconocimiento de importantes hallazgos que, de muchas maneras, fueron los predecesores de la biotecnología actual. En el año 1945, Fleming, Florey y Chain recibieron el Premio Nobel por descubrir que los antibióticos se podían obtener de microorganismos (23). Este fue un vislumbre revolucionario de cómo la biotecnología podía crecer para servir a la terapia farmacológica. En el año 1955, Vincent du Vigneaud recibió el Premio Nobel en Química por la síntesis de los primeros péptidos usando técnicas bioquímicas (oxitocina y vasopresi-na) (24). Esto reveló el interés en técnicas de laboratorio que podrían utilizarse para sintetizar proteínas, y la necesidad de las mismas. En el año 1959, Severo Ochoa y su discípulo Arthur Kornberg, recibieron el Premio Nobel en Medicina y Fisiología por el descubrimiento de los mecanismos de síntesis de los ácidos nucleicos ARN y ADN (25), un evento fundamental que iba a tener consecuencias decisivas para el posterior desarrollo de la ingeniería genética. Un homenaje digno para los farmacólogos, el Dr. Ochoa ocupó la Dirección de Farmacología en la Escuela de Medicina de la Universidad de Nueva York. En el año 1978, Arber, Nathans y Smith recibieron el Premio Nobel en Medicina y Fisiología por el descubrimiento de las enzimas de restricción (26) y, en el año 1980, Berg, Gilbert y Sanger recibieron el Premio Nobel en Química por ocupar el liderazgo en el desarrollo de la tecnología del rADN (27). El logro de Sanger fue particularmente meritorio, debido a que él ya había recibido el Premio Nobel en 1958 por aclarar la estructura de la insulina. Otros logros también merecen reconocimiento; por ejemplo, el descubrimiento de la estruc-tura molecular del ADN por Watson, Crick y el frecuentemente olvidado Wilkins (28) ganadores del Premio Nobel en Medicina y Fisiología en el año 1962. Si revisamos la lista de los premiados en Medicina y Fisiología o Química, más allá de la producción de la proteína usando procedimientos de rADN, podemos observar cuántos otros hallazgos también están directamente relacionados con la biotecnología moderna. Un claro ejemplo se puede apreciar en Köhler, Milstein y Jerne, quienes obtuvieron el Premio Nobel en Medicina y Fisiología en 1984 por proponer y desarrollar la técnica de producción del anticuerpo monoclonal (29). Sin embargo, la síntesis proteica usando el rADN de diferentes especies se ha convertido en la técnica básica para producir fármacos proteicos. Permítanos examinar esta técnica con más detalle (Figura 1).
La producción de moléculas recombinantes es difícil y costosa. La identificación del gen, la elección de la población de células que expresan la proteína, y la purificación de la proteína producida son solo algunos de los complejos pasos necesarios para la biosíntesis de las proteínas recombinantes terapéuticas, que también requiere de monitoreo durante todo el proceso(32). En general, esto es infinitamente más prolongado y complejo que la producción de fármacos mediante síntesis química. Por ejemplo, un producto químico tradicional requiere de 100 a 200 ensayos de control de calidad, mientras que un fármaco biotecnológico puede requerir más de 2000 (incluido el control en el proceso y el análisis del producto) (33). Esta complejidad es extremadamente relevante: variaciones aparentemente insignificantes en el proceso de producción pueden conllevar a cambios estructurales en la molécula final que puede tener un enorme impacto en el comportamiento farmacológico del producto. En el caso de los fármacos biotecnológicos, se dice que el proceso es el producto, debido a que la estructura, la conformación y, por lo tanto, las propiedades farmacológicas del producto final se vinculan estrechamente con los procedimientos aplicados durante la fabricación (34). Esto es comprensible ya que el comportamiento de los organismos vivos está relacionado estrechamente con los cambios ambientales, mucho más que los compuestos derivados de químicos. Esto muestra justamente cuán diferentes son los fármacos biotecnológicos de los fármacos sintéticos, los cuales admiten una mayor flexibilidad en su proceso de producción. Un ejemplo bastante discutido que ilustra la relevancia del protocolo de producción de agentes biotecnológicos es la elección del sistema para la expresión de la proteína recombinante humana. Las células procariotas (bacterias) permiten una producción de proteínas más rápida, debido a que, como se supone que una sola bacteria se divide cada 20 minutos, se formarán mil millones de células en poco más de medio día (se cultivan en tanques capaces de contener miles de litros). Aparentemente, esta es una ventaja de los sistemas procariotas. Por otro lado, las células eucariotas de mamíferos, tales como aquéllas del Ovario de Hámster Chino (CHO), crecen de manera más lenta y, por lo tanto, toman considerablemente más tiempo en producir una cantidad similar del biofármaco. La secuencia de aminoácidos de la proteína resultante en ambos vectores de expresión es la misma, a pesar de que sabemos que las bacterias no pueden glicosilar proteínas (35) (es decir, no pueden añadir carbohidratos, o azúcares; proviene del griego glycos que significa dulce), mientras que las células de mamíferos sí pueden. En el caso de algunas proteínas, la glicosilación es esencial para que éstas sean activas (35-37). Este es un ejemplo extremo de la importancia de elegir el proceso de producción. Otros cambios, a pesar de que son aparentemente poco significativos, son a veces incluso más decisivos para la viabilidad terapéutica de los medicamentos biológicos recombinantes, especialmente en términos de seguridad. Por ejemplo, la formulación del producto final determinará la estabilidad, la conformación y, por consiguiente, la actividad biológica (38,39). A pesar del hecho de que los farmacólogos, en general, aún no están familiarizados con esta metodología, estamos por desarrollar proteínas recombinantes más sofisticadas o, al menos, proteínas que requieren procesos de producción más sofisticados. Ya se encuentran en el mercado proteínas que son creadas uniendo partes de dos o más genes diferentes, los cuales se codifican originalmente para proteínas independientes (proteínas de fusión) (40), y ya existen toxinas vinculadas a un anticuerpo dirigido a un antígeno específico asociado a tumores de superficie celular (inmunotoxina) o toxinas quiméricas (41,42). La situación se torna más complicada, debido a que la recombinación genética para obtener proteínas se puede llevar a cabo en plantas (43) y animales de granja que, en consecuencia, se vuelven transgénicos (la denominada granja farmacéutica). Por ejemplo, en el año 2006, la EMA aprobó una antitrombina-a producida en la leche de cabras en la que se había insertado el gen correspondiente (44). Para concluir sobre este tema, en comparación con las técnicas tradicionales para obtener moléculas proteicas, la metodología del ADN recombinante permite obtener los productos de manera más rápida y en cantidades mayores. Estos productos son más puros, menos susceptibles a contaminación y, si se desea, más similares a la molécula endógena. El éxito de esta técnica tiene una consecuencia inevitablemente aritmética: el crecimiento exponencial en el número de fármacos proteicos que se producen y se espera que lleguen al mercado. Si tomamos fármacos biotecnológicos recombinantes actualmente autorizados, aquéllos que están a la espera de autorización y aquéllos proyectados, el tamaño de mercado previsto es casi inimaginable (45). En 5 años, desde el 2003 al 2008, el mercado global para los fármacos biotecnológicos ha aumentado en más del doble, de US$38 mil millones a US$83 mil millones. Esto, a pesar de que la participación en el mercado de los medicamentos convencionales está disminu- yendo progresivamente a favor de los biomedicamentos. Este es un aviso particularmente relevante para los farmacólogos. Sin embargo, ¿de qué manera la naturaleza de la proteína de un fármaco afecta su comportamiento farmacológico? Farmacología de proteínas terapéuticas recombinantes: Inmunogenicidad y otros aspectosAdemás del proceso de producción, las proteínas recombinantes se diferencian de los fármacos tradicionales sintetizados químicamente de dos maneras básicas: en su estructura química (obviamente proteica) y en la similitud estructural y funcional entre las proteínas recombinantes y moléculas endógenas. Estos aspectos de los fármacos proteicos biotecnológicos afectan considerablemente su compor-tamiento farmacológico y la metodología necesaria para su investigación y desarrollo. La complejidad implicada en el desarrollo y estudio de proteínas terapéuticas, y el impacto de esta complejidad en la farmacocinética, en comparación con productos químicos tradicionales, está ilustrada, por ejemplo, por diferencias en el tamaño. La EPO, por ejemplo, es aproximadamente 170 veces más grande que el ácido acetilsalicílico (otras moléculas son hasta 1000 e incluso 10000 veces más pequeñas en comparación con los anticuerpos monoclonales). El tamaño es importante en la farmacología. Además, a pesar de que se han desarrollado formas que resisten la degradación en los sitios de administración (46), la biodisponibilidad de estos fármacos es generalmente baja, y su distribución y penetración en el tejido son bastante limitadas (47-49). Además, el hecho de que los fármacos copien, con frecuencia, moléculas endógenas significa que el estado fisiopatológico del paciente tiene un considerable efecto en su actividad (47). No solo los farmacólogos requieren conocimientos profundos de la farmacocinética y farmacodinámica de las proteínas terapéuticas para la investigación y desarrollo, sino que la tecnología que circunda estos compuestos es también considerablemente diferente de aquélla que se utiliza, por lo general, en los fármacos sintetizados químicamente. Los fabricantes frecuentemente tienen que diseñar técnicas para evaluar las moléculas de proteína, asegurando que estos métodos distingan entre el medicamento biológico y su contraparte endógena (50), y desarrollar métodos de detección de anticuerpos contra el medicamento biológico. Los farmacólogos, por su parte, requieren nuevos conocimientos técnicos, con la finalidad de ser capaces de investigar fármacos biotecnológicos. La farmacología deberá incluir la formación de los biofarmacólo logos preclínicos y clínicos, debido a que la introducción de estos fármacos desafiará algunos de los conceptos tradicionales de la farmacología. Esto se puede observar, de manera particularmente clara, en la preocupación que surge por los efectos adversos que resultan de las reacciones inmunológicas que generan estos nuevos compuestos, es decir, su inmunogenicidad; anecdótico hasta la fecha en la farmacología de moléculas pequeñas. ¿Por qué la inmunogenicidad se ha convertido repentinamente en un motivo de preocupación? Es fácil entender que las moléculas grandes, complejas y, con frecuencia, inestables, tales como las proteínas terapéuticas y péptidos, tienen una mayor probabilidad de expresar regiones que se comportan como epítopos (estructuras contra las cuales reacciona el sistema inmunológico). La habilidad de estos fármacos proteicos de iniciar la respuesta inmunológica se conoce como inmunogenicidad. Es incluso más fácil entender la tendencia de las proteínas de volverse inmunogénicas si, como a veces es el caso, estas moléculas de proteína no son exclusivamente de naturaleza humana. Esta es la razón por la que el posible beneficio terapéutico de la administración de anticuerpos monoclonales murinos se vio ensombrecido por la reacción inmunológica que ellos generaron cuando se identificaron como extraños. Estos anticuerpos se conocen como anticuerpos humanos anti-ratón (HAMA). Esto desencadenó el desarrollo de anticuerpos monoclonales quiméricos humano-murinos (con el sufijo -ximab, por ejemplo, rituximab), seguidos por anticuerpos humanizados con un mayor contenido humano (con el sufijo -zumab, por ejemplo, trastuzumab), e incluso los anticuerpos completamente humanos disponibles actualmente (con el sufijo -umab, por ejemplo, adalimumab). A pesar de que estos agentes contribuyen en gran medida a reducir la inmunogenicidad, ellos no solucionan el problema completamente (51). Esto no debería ser una sorpresa, ya que incluso las proteínas recombinantes terapéuticas con una secuencia de aminoácidos que es idéntica a aquélla de una contraparte natural, no están exentos de llevar determinantes antigénicos. No es fácil explicar por qué una proteína terapéutica recombinante puede ser inmunogénica, a pesar de la similitud de dicha estructura primaria. No obstante, se pueden hacer algunos comentarios (52,53). Las proteínas recombinantes terapéuticas son compuestos tridimensionales y estructuras cuaternarias a veces complejas, cuyo plegamiento genera una forma espacial que puede ser subóptima y frecuentemente inestable (54). Su inestabilidad durante el almacenamiento puede ser ocasionalmente suficiente para generar nuevas estructuras, tales como agregados de proteína o productos que resultan de la degradación química, que actúan como epítopos. Además, el proceso de purificación de proteínas durante la producción frecuentemente lleva impurezas procedentes de los vectores de expresión (por ejemplo, porciones de ADN de genes bacterianos), y estas impurezas pueden provocar inmunogenicidad (53). La inmunogenicidad no se percibiría como un elemento que se debe evitar si no tuviera consecuencias clínicas. Los medicamentos biológicos pueden, y comúnmente lo hacen, generar anticuerpos contra los fármacos que no tienen un impacto clínico, pero estos mismos anticuerpos podrían provocar que falle la terapia o se desarrollen reacciones adversas severas (55,56). A pesar de que no son comunes las reacciones adversas severas, un episodio que ocurrió al final del siglo pasado e inicios de este, nos advirtió acerca de la necesidad de evaluar las posibles consecuencias clínicas de un caso hipotético de inmunogenicidad. Tal como se manifestó anteriormente, la rhEPO fue una molécula revolucionaria que se comercializó en los años 80. Ha evitado muchas transfusiones, ha elevado la calidad de vida de pacientes con insuficiencia renal crónica, y ha reducido al mínimo muchos de los efectos adversos de la quimioterapia en pacientes con cáncer (19,57). Una de las compañías que lanzó la rhEPO introdujo una variación aparentemente poco significativa -cambió la albúmina por polisorbato 80- en su formulación. A partir de allí, se empezaron a observar reacciones adversas muy severas y a veces, aunque escasas, fatales en pacientes tratados con un tipo específico de jeringa prellenada (58). Se han propuesto diversas hipótesis para explicar estas reacciones (59). Lo más recomen-dable parecía ser la formación de agregados o lixiviados que potencian la producción de la neutralización de los anticuerpos anti-EPO que reconocieron no solo el fármaco, sino también la propia eritropoyetina del paciente (60). Los anticuerpos que reaccionaron con la eritropoyetina endógena, rompiendo de ese modo su tolerancia inmunológica, causaron un tipo de anemia súbita conocida como aplasia pura de células rojas (PRCA), que tuvo consecuencias fatales. A pesar de que los problemas destacados por este caso parecen haberse superado ahora, la EMA ha intensificado sus requisitos de seguridad para la aprobación de medicamentos biológicos. Una situación similar se observó con la trombopoyetina recombinante (61), a pesar de que afortunadamente ésta se detectó durante el desarrollo clínico. Finalmente, independientemente de la severidad de las reacciones adversas, la inmunogenicidad puede provocar fracasos en la terapia debido a que los anticuerpos que interactúan con fármacos proteicos externos los neutralizan atenuando o bloqueando su actividad (62); un fenómeno observado, por ejemplo, en las preparaciones del IFN y el Factor VIII (63). Demostrar la existencia de anticuerpos neutralizantes durante el desarrollo de un medicamento biológico es una importante advertencia para la compañía farmacéutica o entidades regulatorias, que a veces están en contra de su desarrollo posterior. En abril de 2008, la EMA, la agencia a cargo de la autorización de los fármacos biotecnológicos en Europa, publicó directrices definitivas junto con recomendaciones que los laboratorios farmacéuticos deben adoptar para evaluar la inmunogenicidad (64). Biosimilares, no biogenéricos Los requisitos generales para el desarrollo preclínico y clínico de un medicamento biotecnológico innovador son fundamentalmente los mismos que aquéllos de un fármaco sintético, pese al hecho de que, en promedio, la inversión necesaria es mucho mayor para el biofármaco. A pesar de que podríamos enfocarnos en aspectos específicos del desarrollo de los fármacos (por ejemplo, la necesidad de evaluar la inmunogenicidad en el caso de los medicamentos biológicos), aquí examinamos por qué es más exigente hacer una copia de un medicamento biológico que de un fármaco sintetizado químicamente (pequeño), un denominado genérico. Ya hemos visto cómo, incluso, una leve variación en el proceso de producción de un medicamento biológico puede modificar las características químicas del producto final (es decir, el proceso es el producto), y que esto podría, a su vez, conllevar a cambios en el comportamiento biológico de la molécula (65) y, en consecuencia, a cambios en su seguridad y/o eficacia (66). Estas consecuencias se deben tomar en cuenta, por ejemplo, cuando una compañía pretende modificar el protocolo de producción de un fármaco innovador que ya se ha lanzado al mercado (67); sin embargo, también se deben considerar al intentar producir una copia de un medicamento biotecnológico cuya patente ha expirado (68,69). La complejidad de las proteínas terapéuticas y sus técnicas de producción con base en organismos vivos, así como los requisitos de confidencialidad de las compañías farmacéuticas, hacen imposible que otra compañía produzca una copia estructuralmente idéntica del fármaco innovador (70). Ligeras diferencias en la composición podrían comprometer la seguridad y eficacia del fármaco, debido a que no es suficiente para que la secuencia del aminoácido de la copia sea la misma. Ya que no es posible producir dos biofármacos idénticos ni garantizar completamente su estructura molecular tridimensional debido, entre otras cosas, a modificaciones postranslacionales que conllevan a la microheterogeneidad de aquellos medicamentos, los criterios del desarrollo para un fármaco biotecnológico deben ser más estrictos que aquéllos requeridos para un genérico, para el cual solo la bioequivalencia farmacocinética es generalmente suficiente para probar un comportamiento terapéutico idéntico (71). En consecuencia, para hacerlos equivalentes, la copia de un medicamento biotecnológico debe demostrar ser tan eficaz y seguro como el fármaco de referencia innovador en ensayos clínicos comparativos que evalúen ambos aspectos. Asimismo, se deben realizar comparaciones preclínicas y es obligatoria la información de seguridad poscomercialización de la copia. Nuevamente, en cuanto a las normas de la EMA, los laboratorios biofarmacéuticos que producen innovadores tratamientos biotecnológicos también necesitan demostrar que no existen cambios en la eficacia y seguridad cuando se aplica un cambio importante del proceso de fabricación en el producto innovador. La EMA utiliza el término biosimilar o medicamento biológico similar para la copia autorizada de un medicamento biológico, por consiguiente, biotecnológico producido de conformidad con requisitos específicos de calidad, eficacia y seguridad (72). Con la finalidad de evitar confusiones, no se deberá utilizar el término biogenérico debido a que sus vías reguladoras correspondientes para obtener la autorización son diferentes. En el año 2005, la EMA emitió directrices sobre los biosimilares (73,74), convirtiéndola, en consecuencia, en la única entidad regulatoria hasta la fecha que ha propuesto una vía abreviada para su desarrollo (Japón y Canadá están en camino). Veremos cuál será la posición de la FDA con respecto a los biosimilares o follow-on biologicals (FOB) como supuestamente se les llamaría en los Estados Unidos. Desde la aprobación en Europa de dos biosimilares de somatotropina en el año 2006, la EMA ha autorizado 13 biosimilares (de hecho, realmente seis diferentes moléculas). Antes de emitir las directrices, se había rechazado una copia de un producto biotecnológico, y al menos otro fue retirado, más adelante, de la evaluación de la EMA por el promotor. El exigente ejercicio de comparar un biosimilar con el fármaco innovador requiere más tiempo y esfuerzo, así como una inversión considerablemente mayor (entre 5 y 100 veces), que los requeridos para el desarrollo de un medicamento genérico. A pesar de que se prevé que el precio de los biosimilares también lleve a los innovadores a aplicar un descuento en sus productos, las exigentes normas supuestamente no permitirán descuentos cerca a aquéllos alcanzados en los genéricos. Se espera que el mercado de los biosimilares crezca en los siguientes años. Afortunadamente, cubren una necesidad social en un mundo donde los gastos de asistencia sanitaria están creciendo a una velocidad exponencial. Sin embargo, la inversión necesaria ha generado aprehensión y dudas con respecto a la viabilidad financiera de estos agentes. Solo queda por ver, por lo tanto, con qué rapidez la industria de los biosimilares se desarrollará en Europa y el resto del mundo. La creciente incertidumbre en muchas compañías sobre la posibilidad de recuperar la inversión original se agrava por el hecho de que, en algunos países, la legislación nacional impide que los químicos farmacéuticos realicen la sustitución automática a fármacos biotecnológicos (a diferencia de los innovadores fármacos sintéticos y genéricos) (75). Es importante destacar que esta política de substitución no automática se aplica en algunos países a cualquier fármaco biotecnológico, también productos innovadores, no específicamente a biosimilares. Esto significa, por ejemplo, que el farmacéutico no puede entregar una EPO innovadora si el médico prescribe otro innovador (existe más de una EPO original en el mercado). Lo mismo se aplicaría a los dos IFNß-1a originales actualmente en el mercado para la esclerosis múltiple. Por lo tanto, debemos decir que los biosimilares son fármacos que están garantizados terapéuticamente tanto como los correspondientes biomedicamentos originales en términos de calidad, eficacia y seguridad, y que, una vez que la molécula haya sido aprobada por la EMA, ya no es una copia sino otro medicamento biotecnológico. Algunos argumentan que incluso un biomejorado (biobetter) se puede producir en algunas ocasiones en las que se utilizan técnicas modernas para su producción. Finalmente, permítannos no olvidar que una copia de un biomedicamento que no ha ingresado en la vía del EMA para su autorización y, sin embargo, se ha introducido en mercados no europeos, no se deberá llamar un biosimilar. Conclusión. Medicamentos biotecnológicos (tanto innovadores como biosimilares): ambos una necesidad y un desafío A partir de lo que se ha discutido anteriormente, está claro que los medicamentos biológicos no son simplemente una nueva familia de fármacos, como lo fueron, por ejemplo, los inhibidores de la enzima convertidora de la angiotensina, los inhibidores de la bomba de protones, o los antagonistas del receptor de leucotrienos en su día. Esta nueva generación de moléculas terapéuticas anuncia una nueva era en farmacología, una que requerirá que los farmacólogos adquieran nuevas habilidades para desarrollar y manejar estas moléculas. Estos nuevos fármacos han abierto, por lo menos, una nueva rama completa de la farmacología. Tal vez sea tiempo de empezar a utilizar el viejo término de biofarmacología, ahora en desuso, con un diferente significado. Podríamos, incluso, llegar más lejos. Si aceptamos, como lo han propuesto algunos pensado-res, que una ciencia utiliza sus propios recursos para explicarse a sí misma, la biofarmacología podría convertirse en una ciencia emergente en la intersección de varias otras disciplinas. Más que un prometedor desarrollo para el futuro, los agentes biotecnológicos son una realidad tangible. Sin embargo, como la mayoría de los nuevos productos, su desarrollo y comercialización plantean muchas preguntas. Depende de los fabricantes, investigadores, médicos, farmacéuticos y regula- dores coordinar esfuerzos para ayudar a responder estas preguntas, con la finalidad de asegurar que los tratamientos sean tanto seguros como eficaces. Garantizar la seguridad de los fármacos y el acceso de los pacientes a estos importantes fármacos deberá ser el máximo objetivo de todos aquellos actores. Los intereses partidistas significan que muchas preguntas se dirigen hacia áreas específicas de las normas pertinentes. Afortunadamente, la naturaleza de estas normas en Europa sirve como una garantía para los pacientes. Este es un enfoque conveniente, desde el punto de vista de la EMA, ante la incertidumbre sobre una gran cantidad de fundamentos científicos y médicos subyacentes de muchos de estos fárma-cos, y la ignorancia acerca de los mismos. ¿Se justifica que el farmacéutico no pueda sustituir automáticamente los agentes biotecnológicos en algunos países? ¿Justifican pocos casos de inmunogenicidad fatal tales normas estrictas? Dados estos requisitos, ¿alcanzará la participación en el mercado de los biosimilares aquélla del mercado genérico? ¿Desarrollará la Administración de Alimentos y Fármacos (FDA) un conjunto de normas similares como aquél de la EMA con respecto al FOB? ¿Competirán los biosimilares solo en el precio? ¿Será posible la eficaz administración entérica de proteínas terapéuticas? ¿Se utilizarán las plantas (si bien transgénicas) nuevamente como la fuente principal de los fármacos? Solo el tiempo proporcionará respuestas a éstas y otras preguntas. Por ahora, solo el horizonte es visible. Pero sabemos que algo, probablemente casi todo, se encuentra más allá de él. Referencias bibliográficas sugeridas
(1) Director del Departamento de Farmacología, Terapéutica y Toxicología de la Universidad Autónoma de Barcelona. Consultor en desarrollo y comercialización de biosimilares en mercados específicos.(2)Investigadora posdoctoral senior en las instalaciones de investigación del Hospital Clínico de Barcelona (España).(* )Publicado con permiso del autor y del editor de la Revista Journal of Generic Medicines. |
||