|
|
| Características clínicas de la parálisis supranuclear progresiva en el Instituto Nacional de Ciencias Neurológicas Luis Torres Ramirez(1),Mirla V. Villafuerte Espinoza (2),Erik Guevara Silva (3),Carlos Cosentino Esquerre (4) ResumenIntroducción: La Parálisis Supranuclear Progresiva (PSP) es la segunda causa de parkinsonismo plus más común. La parálisis de la mirada vertical y la inestabilidad postural con caídas en el primer año de inicio de síntomas son claves para el diagnóstico. El objetivo de este estudio fue hacer una descripción clínica de una población peruana con PSP. Población y Métodos: Estudio descriptivo y retrospectivo de pacientes con PSP atendidos desde 1996 al 2010, según la base de datos del Departamento de Enfermedades Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas. Resultados: Dieciséis pacientes cumplieron los criterios de inclusión, 11 (68.8%) con PSP probable y 5 (31.2%) con PSP posible. Se evidenció un predominio masculino (M:F = 7:1). La edad promedio de inicio de síntomas fue de 58.4 años. Durante la primera evaluación los síntomas más frecuentes fueron la rigidez, bradicinesia e inestabilidad postural, mientras que en el último control todos presentaron limitación de la mirada vertical. Los estudios de neuroimagen se realizaron en 13 pacientes, de los cuales 12 mostraron alteraciones. Conclusiones: Se presentan las características clínicas de la PSP en Perú. Nuestro estudio reveló un predominio del sexo masculino similar a otros estudios; sin embargo la edad de inicio de enfermedad es menor comparado a otras latitudes. Palabras clave: Parálisis de la Mirada Vertical, Parálisis Supranuclear Progresiva, Parkinsonismo, Síndrome de Steele-Richardson-Olszewski, Parálisis de la Mirada Vertical. AbstractIntroduction: Progressive supranuclear palsy (PSP) is the second most common parkinsonism. Vertical gaze palsy and postural instability with falls in the first year of symptoms onset are key for diagnosis. Patients and Methods: A descriptive and retrospective study included every patient diagnosed as PSP since 1996 at 2010 from Neurodegenerative Disease Department database of the Instituto Nacional de Ciencias Neurológicas. Results: Sixteen patients fulfilled the diagnostic criteria, 11 (68.8%) with probable PSP and 5 (31.2%) with possible PSP. A male predominance (M:F = 7:1) was evidenced. The mean age at onset of disease was 58.4 years. Rigidity, bradykinesia and postural instability were the most frequent symptoms in the first evaluation, whereas vertical gaze palsy was present in all patients in the last consultation. Neuroimaging was done in 13 patients, 12 of which were abnormal. Conclusions: The clinical characteristics of PSP in Perú are presented. Our study revealed a predominance of male sex similar to other studies; however, the age at onset of disease is minor compared to other latitudes. Key words: Parkinsonism progressive supranuclear palsy, Steele-Richardson-Olszewski syndrome, vertical gaze palsy. Introducción La Parálisis Supranuclear Progresiva (PSP) o Síndrome de Steele-Richardson-Olsewski es el más común de los trastornos parkinsonianos luego de la Enfermedad de Parkinson(1-2) y al mismo tiempo, es un trastorno neurodegenerativo progresivo de origen desconocido(3); sin embargo, podría estar relacionado con el medio ambiente y factores genéticos(4-5). Independientemente de la causa primaria, las investigaciones han relacionado la aparición de los ovillos neurofibrilares con la disfunción mitocondrial y el estrés oxidativo (6). La PSP fue establecida por primera vez por Steele, Richardson y Olszewski en 1964, cuando ellos describieron nueve pacientes con síndrome neurológico caracterizado por parálisis supranuclear progresiva de la mirada, rigidez axial, síndrome parkinsoniano, y parálisis pseudobulbar. Seis de los casos fueron examinados histológicamente y encontraron una degeneración neurofibrilar extensa particularmente en el estriado, globo pálido medial, núcleo subtalámico y la sustancia negra (1). Esta enfermedad se asocia a anormalidades en los microtúbulos con una hiperfosforilación anormal de las proteínas Tau; la agregación de estas proteínas son responsables de la degeneración neurofibrilar por ello se la clasifica como una taupatía junto con la enfermedad de Alzheimer, enfermedad de Pick, degeneración corticobasal, demencia frontotemporal, entre otras (3) . La prevalencia varía entre 1.39 y 4.9 casos por 100,000 habitantes, con una incidencia desde 0.3 a 1.1 por 100,000 al año (7). En el Reino Unido revelaron que la enfermedad es más común de lo que se había considerado previamente, con una prevalencia de 6.5 casos por 100 000 personas (8). En la isla de Guadalupe la incidencia de PSP es 14 / 100, 000, parecía ser una asociación entre el consumo a largo plazo de algunas frutas tropicales y té de hierbas que contiene tetrahydroisoquinolonas (9). En 1996 se publicaron los primeros criterios diagnósticos validados internacionalmente (10). El 2003 estos criterios fueron redefinidos por un grupo de expertos y se agregaron las categorías de PSP posible, probable o definido, esta última requiere confirmación patológica (11), considerándose a la parálisis de la mirada vertical y la inestabilidad postural con caídas en el primer año como síntomas claves para el diagnóstico (5).
Actualmente no existen publicaciones sobre la PSP en nuestro país, constituido por una población étnica y culturalmente diversa; el propósito del estudio fue analizar el espectro clínico de un grupo de pacientes con diagnóstico de PSP que fueron atendidos en nuestra institución en los últimos 15 años. Material y método Este estudio descriptivo de corte longitudinal y retrospectivo incluyó a todos los pacientes identificados que fueron internados y tratados como PSP desde enero de 1996 y diciembre del 2010 en el Departamento de Investigación, Docencia y Atención Especializada en Enfermedades Neuro-degenerativas del Instituto Nacional de Ciencias Neurológicas. Se revisaron las historias clínicas de cada paciente para obtener las características clínicas, demográficas y resultados de exámenes auxiliares los cuales fueron anotados en una ficha estructurada y precodificada. La categoría diagnóstica se determinó de acuerdo a los criterios diagnósticos de consenso. Criterios de inclusión El estudio incluyó a todo paciente con PSP diagnosticada por neurólogo con experiencia en enfermedades neurodegenerativas (L.T . C.C.) y en base a los criterios diagnósticos mencionados arriba. Criterios de exclusión
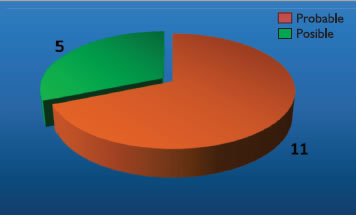
•Pacientes que presentaron inicialmente un cuadro clínico sugestivo de PSP pero que luego fueron manifestándose síntomas que sugerían otro tipo de parkinsonismo neurodegenerativos o cuando se descubrió en los exámenes de imágenes que se trataba de un parkinsonismo secundario. Análisis estadístico Se utilizó el programa estadístico SPSS 17. Los resultados se expresaron mediante pruebas estadísticas descriptivas, gráficos, cuadros de distribución de frecuencias y porcentajes para la determinación de la magnitud y características de la problemática en estudio. Toda información recogida se consideró estrictamente confidencial y de manejo exclusivo por parte del grupo investigador. Los resultados son expresados de manera estratificada o global sin identificación de personas. Resultados Revisamos los registros médicos de 18 pacientes con diagnóstico de PSP, de los cuales 16 cumplían con los criterios de inclusión. Del total de pacientes, 11 (68.8%) cumplieron los criterios para PSP probable y 5 (31.2%) se catalogaron como PSP posible.
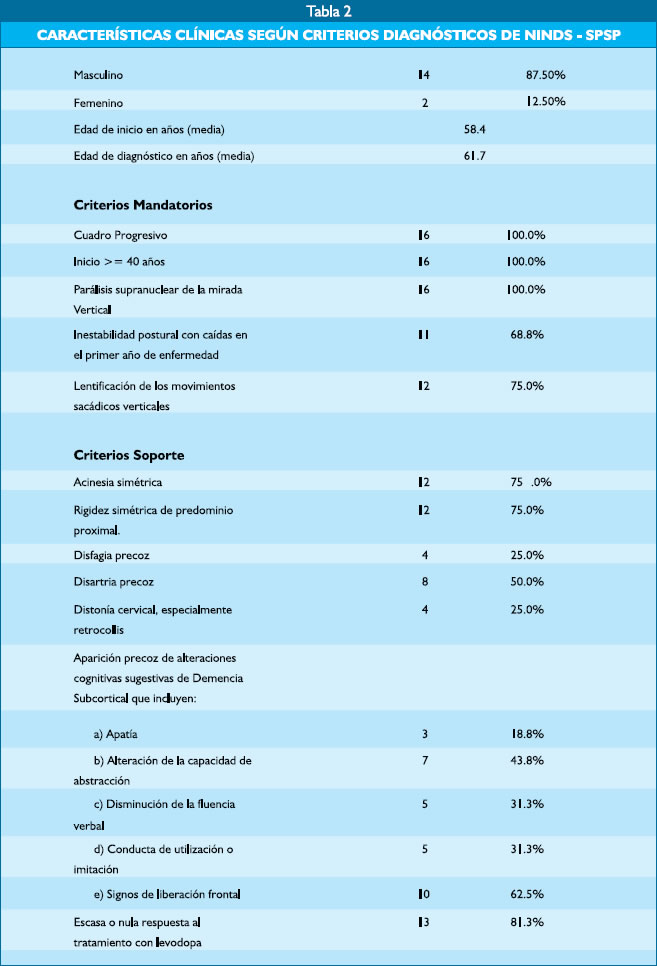
Encontramos 14 varones (87.5%), 2 mujeres (12.5%). El promedio de edad de inicio de los síntomas fue de 58.4 años (rango, 40 a 72 años) y al momento del diagnóstico 61.7 años (rango, 41 a 78 años). Las principales características clínicas se describen en la tabla 2.
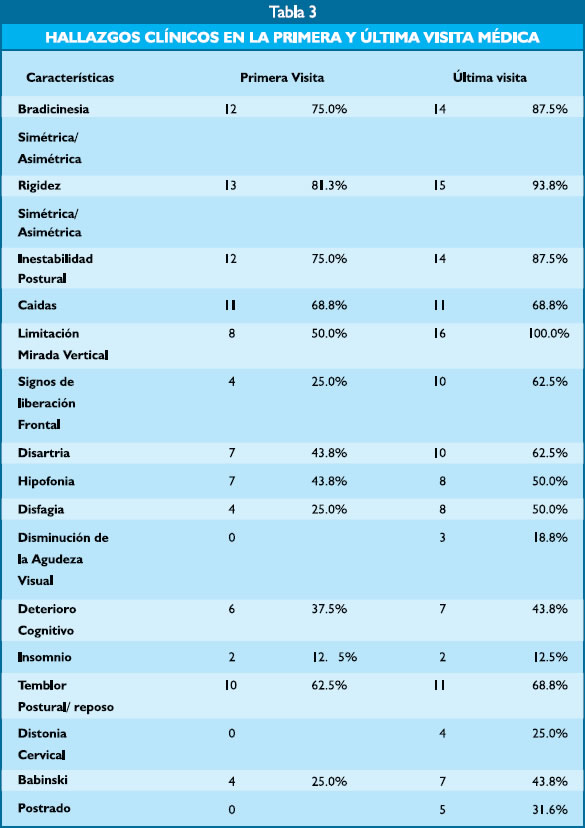
Los pacientes asistieron al instituto con un tiempo de inicio de enfermedad promedio de 3.31 años (rango 1-8 años). Durante la primera evaluación médica los síntomas más frecuentes fueron la rigidez en 13 pacientes (81.3%), bradicinesia e inestabilidad postural en 12 pacientes (75%), mientras que en el último control médico todos los pacientes presentaron limitación de la mirada vertical y 5 pacientes habían llegado a la postración (31.6%). Tabla 3.
Cuatro pacientes no presentaron ninguna comorbilidad, en el resto (75%) las condiciones comórbidas más comunes fueron: HTA (4 pacientes), diabetes mellitus 2 (3), patología prostática (3), artritis reumatoide (2), neumonía (1), enfisema pulmonar (1) insuficiencia renal crónica (1), osteoartrosis (2), anemia crónica (1). Todos los pacientes recibieron Levodopa durante la enfermedad, en algunos se llegó a una dosis máxima de 1000 mg día, de los cuales 3 pacientes (18.7%) refieren algún grado de mejoría subjetiva al inicio de la enfermedad, no se evidenció discinesia relacionada al tratamiento con levodopa, 13 (81.3%) pacientes no refieren mejoría al tratamiento, además utilizaron selegilina, biperideno, bromocriptina, pramiprexol, pergolide, ácido valproico, con escasa eficacia. Los diagnósticos inicialmente planteados fueron enfermedad de Parkinson en 5 pacientes (31.3%), atrofia multisistémica en 2 pacientes (12.5%), degeneración corticobasal en 1 paciente (6.3%) y el resto fue considerado como parkinsonismo sin especificar. Los estudios por neuroimágenes fueron realizados en 13 pacientes (81.3%), de los cuales 7 fueron por resonancia magnética cerebral (RMN) y 6, por tomografía axial computarizada (TAC). De los pacientes que se hicieron RM se informaron: atrofia cortico-subcortical bilateral y microinfartos en sustancia blanca (4 pacientes), atrofia cortico subcortical (1 paciente), discreto aumento de volumen de espacios sub-aracnoideos y ventrículos (2 pacientes). En los que se realizaron tomografía se informaron como atrofia cerebral (2 pacientes), atrofia tectal (1 paciente), atrofia cerebral y cerebelosa simétrica (1 paciente), atrofia cortico subcortical e infarto lacunar frontal derecho (1 paciente), normal (1 paciente). Discusión Encontramos 16 pacientes que cumplieron con los criterios clínicos diagnósticos de NINDS-SPSP (11). Los crite- rios propuestos para PSP Probable tiene alta especificidad (100%), pero con menos sensibilidad al comienzo de la enfermedad, mientras que los criterios para PSP Posible fueron más sensibles (83%) en la visita de los primeros 3 años después del inicio, pero son menos específicos (7) . En un reporte de 143 casos de parkinsonismos del Reino Unido hubo 19 casos confirmados por patología de PSP, los criterios clínicos utilizados tuvieron un valor predictivo positivo, sensibilidad y especificidad para el diagnóstico de PSP de 80,0%, 84,2% y 96,8%, respectivamente (12) . Se evidenció un predominio masculino (M:F, 7:1) mayor a lo reportado por Mestrinelli et al, quienes estudiaron en forma similar a 16 pacientes (10 varones y 6 mujeres, M:F , 1.6:1) en una población de Brasil (13). Aunque ha habido informes de un predominio de los varones con esta patología (1,14-15), en las publicaciones recientes refieren que ambos sexos son igualmente afectados (5,7,16) . La edad promedio de inicio de enfermedad (58.4 años) fue menor a la referida por este autor (64.7 años) y en diferentes series de casos como lo reportado en USA y Reino Unido, el promedio de edad se encontró entre los 60 y 66 años (7,16).
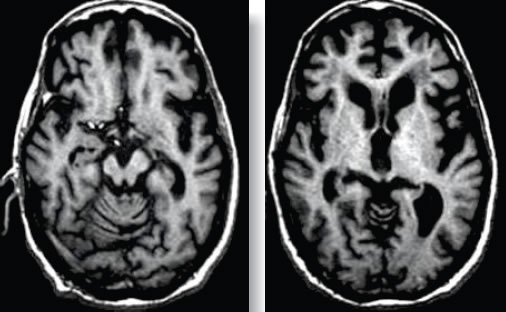
La primera visita ocurre en un promedio de 3.31 años casi similar a lo comunicado por Litvan (3.7 años) (10). En la primera visita los pacientes con PSP tuvieron en orden de frecuencia síntomas de rigidez , bradicinesia, inestabilidad postural, caídas, temblor, limitación de la mirada vertical, disartria, hipofonía; por su parte Litvan reportó como síntomas más frecuentes a la inestabilidad postural y caídas, seguido por disartria y limitación mirada vertical (10) . En la última visita la limitación de la mirada vertical estuvo presente en todos los pacientes, seguido de la rigidez, bradicinesia, inestabilidad postural, caídas y temblor. A diferencia del trabajo de Litvan(10)donde encontramos que todos sus pacientes mostraron inestabilidad postural y bradicinesia, seguido de rigidez axial y disfonía. Dentro de las comorbilidades la más frecuente fue la hipertensión arterial (25%), menor a lo encontrado en el trabajo de Dubinsky et al, 19 de 58 pacientes (32.8%) con PSP (17) y el trabajo de Ghika et al, 34 de 42 pacientes (81%)(18). Sin embargo muchas series patológicas no han comentado una coexistencia significativa de enfermedad de pequeño vaso (19). La escasa o nula respuesta al tratamiento es considerado uno de los criterios de soporte, para el diagnóstico de PSP, en nuestro trabajo se evidenció algún tipo de mejoría en un pequeño porcentaje, pero mayor a lo reportado en el trabajo de Mestrinelli (13) et al. y de Nath et al. (8). La mayoría de investigadores sostienen que un gran número de los pacientes con PSP no responden a la levodopa o a alguna terapia neurotransmisora específica (20-21). La buena respuesta a la terapia con Levodopa va en contra del diagnóstico de PSP; la razón de la ineficacia de las drogas está relacionada con la naturaleza degenerativa amplia que afecta varios núcleos en esta enfermedad (22-24) . En cuanto a los diagnósticos planteados inicialmente, en el 31.3% se consideró como enfermedad de Parkinson y a descartar atrofia multisistémica en 2 pacientes (12.5%) y degeneración corticobasal en 1 paciente (6.3%). Esto demuestra que el diagnóstico sigue siendo un reto sobretodo en las primeras etapas de la enfermedad o si se presenta con variantes fenotípicas (5,8). En ausencia de biomarcadores, los estudios de neuroimágenes señalan ciertas características en la TAC y RMN cerebral orientadores de PSP, aunque en etapas moderadas a avanzadas(25); en la RMN se encuentra atrofia de mesencéfalo, denominada como el signo del Colibri (5,26) o signo de la silueta del pingüino que de perfil parece un pingüino de pie con una cabeza pequeña y cuerpo grande (27), otros signos son incremento del tercer ventrículo, y atrofia del pedúnculo cerebeloso superior. Sin embargo, una limitación de los exámenes de resonancia magnética es que tales alteraciones descritas se detectan en etapas avanzadas de la enfermedad, y no ayudan para el diagnóstico precoz (5,25). Figura 1. Los estudios por neuroimágenes ya sea por RMN o TAC en nuestro trabajo reportaron menos de la mitad de casos con atrofia cerebral y secuela de eventos isquémicos. Es posible que durante la revisión no se hayan incluido muchos pacientes que aún están en estudio sin un diagnóstico probable o posible, o considerados como enfermedad de Parkinson u otro tipo de parkinsonismo y que posteriormente la evolución nos diga que se trata de PSP como sucedió inicialmente con varios de nuestros pacientes. Sin embargo, el estudio de López et al confirma que los criterios de NINDS – SPSP tienen excelente especificidad, además la inestabilidad postural con caídas en el primer año de inicio de enfermedad junto con la parálisis supranuclear de la mirada vertical tiene un gran valor diagnóstico discriminatorio cuando se compara la PSP con otros parkinsonismos (28). Esta revisión retrospectiva se basó en los últimos criterios diagnósticos de consenso, ninguno tuvo confirmación patológica ya que muchos de ellos dejaron de asistir a sus controles. Los exámenes auxiliares así como el tratamiento no fueron aplicados en forma estandarizada. El pequeño número de pacientes de una sola institución no permite establecer la verdadera prevalencia de esta enfermedad. Un estudio prospectivo y multicéntrico sería idóneo para definir la historia natural, respuesta al tratamiento y pronóstico de los pacientes con PSP. Esta es la Primera descripción en nuestro país de una serie de pacientes con PSP, evaluados en un centro neurológico de referencia, utilizando los criterios de consenso de diagnóstico. Nuestro estudio reveló predominio del sexo masculino, y el tiempo promedio de la primera visita médica similar a otras publicaciones; mientras que la edad de inicio de enfermedad fue menor a lo reportado en otras publicaciones; la inestabilidad postural y caídas coinciden con la literatura entre los síntomas que se presentan en la primera visita médica; así como la rigidez, bradicinesia e inestabilidad postural entre los síntomas que se presentan en la última visita médica. Referencias Bibliográficas
1 Médico Neurólogo, Departamento de Enfermedaes Neurodegenerativas del Instituto Nacional de Ciencias Neurológicas. Lima-Perú. 2 Médico Residente de Neurología del Instituto Nacioonal de Ciencias Neurológicas. Universidad Nacional Mayor de San Marcos (UNMSM) Lima-Perú. 3 Médico Neurólogo. Ex Residente del Instituto Nacional de Ciencias Neurológicas. Universidad Nacional Mayor de San Marcos(UNMSM) Lima-Perú. |
||||||||