|
|
| Estudio genético de la hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa en pacientes peruanos y sus familiares
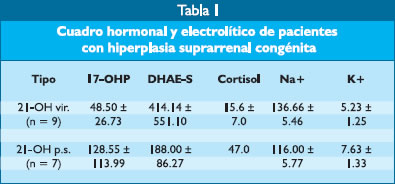
Juan Falen Boggio (1), Carlos Del Águila Villar(2,3),Rómulo Lu de Lama (2),Jorge Chirinos Rodríguez (4),Atsushi Mikami (5) Introducción Antecedentes: La deficiencia de 21-hidroxilasa es la causa de cerca del 95% de casos de hiperplasia suprarrenal congénita y en nuestro medio es responsable de 1 de cada 3000 consultas en el Instituto Nacional de Salud del Niño. El gen que codifica la producción de enzima 21OH es el CYP21B está localizado en el brazo corto del cromosoma 6. Objetivos: Conocer la frecuencia de las principales mutaciones en pacientes peruanos con las formas clásica y no clásica de HSC con deficiencia de 21-hidroxilasa, así como las deleciones del gen CYP21B. Materiales y pacientes: La técnica utilizada para el estudio genético fue la de la polymerase chain reaction (PCR) y se analizaron las mutaciones y delecciones más frecuentes encontradas tanto en Japón como en los EE.UU. de NA. Se han estudiado 20 pacientes que fueron diagnosticados y seguidos en el Servicio de Endocrinología del Instituto de Salud del Niño, 13 padres, 13 madres y 5 hermanos para el estudio genético. Cuando el padre estaba ausente se estudió un familiar de la línea paterna de sexo masculino abuelo o tío, en nuestro estudio se hizo con un abuelo paterno. Resultados: De los 20 pacientes estudiados, 11 fueron de sexo femenino y 9 de sexo masculino; de otro lado, 16 pacientes eran portadores de la variedad perdedora de sal y 4 de la forma virilizante. Tres hermanos de los pacientes no estuvieron afectados y 2 presentaron alteraciones genéticas y cuadro clínico (nacieron cuando se realizaba este estudio). Los pacientes portadores de HSC en el Perú presentan en la variedad perdedora de sal como mutaciones más frecuentes la I172N (7,42%), la P30L (7,42%) y la R356W (7,42%), 7,42% corresponden a deleciones y 21,4% a alteraciones genéticas no detectadas con las técnicas utilizadas; en el caso de la variedad virilizante simple 14,28% de mutaciones corresponden al alelo V28IL, 52,14% a deleciones y 28,57 a alteraciones genéticas no detectadas con la técnica empleada. Conclusiones: Existe una frecuencia de mutaciones alélicas similares a aquellas que se encuentran en la población japonesa, posiblemente por el origen asiático del poblador peruano. Algunas alteraciones genéticas no determinadas corresponderían a mutaciones nuevas o a no analizadas. Palabras clave: Hiperplasia suprarrenal congénita, deficiencia de 21-hidroxilasa, genética. Abstract Background: Congenital adrenal hyperplasia (CAH) in 95% of cases is due to 21-hydroxylase deficiency and of one of every 3000 consultations in the Instituto Nacional de Salud del Niño in Lima, Peru. Deficiency of 21OH is related to mutations in gene CYP21B, located in chromosome 6p. Objectives: To assess the frequency of the main disorders of the CYP 21B gene in peruvian patients with classic and non classic CAH due to 21-hydroxylase deficiency. Methods: 20 patients with CAH of the Endocrinology unit of the IESN were studied. Also, 13 fathers, 13 mothers and 5 brothers where studied. In one case, the paternal grandfather was studied in absence of the father. Polymerase chain reaction (PCR) technique was used in order to determine the mutations and deletions of the samples. The results were compared with those of similar studies in Japan and USA. Results: The frequency by sex in the 20 patients was: 11 females and 9 males. 16 patients had salt loss- CAH and 4 had simple virilizing CAH. Gene disorders and clinical characteristics of CAH were detected in 2 siblings (both were born during the study). The most frequent allele mutations found in salt loss-CAH were I172N (7,42%), P30L (7,42%) and R356W (7,42%). Deletions were detected in 7,42% of cases and unknown gene disorders in 21,4%. In the virilizing CAH cases, the most frequent allele mutation was V281L (14,28), deletions were present in 52,14% and were detected in 28,57% of cases the gene anomaly could not be detected. Conclusions: The frequency of allele mutations in CAH was similar to those found in Japan and USA. The unknown gene disorders may be mutations the gene anomaly could not be detected. Key words: Congenital adrenal hyperplasia, 21-hydroxylase deficiency, genetics. Introducción La glándula suprarrenal histológicamente consta de tres capas bien definidas y posee zonación funcional; la zona glomerulosa produce mineralocorticoides, la fascicular gluco-corticoides y la reticular andrógenos. El colesterol es transformado a través de procesos multienzimáticos en los principales esteroides producidos por dichas zonas. Las zonas fascicular y reticular se encuentran gobernadas por la ACTH y la zona glomerular por el sistema renina-angiotensina y las variaciones de los electrolitos, en especial el potasio (1,2). Cualquiera de las enzimas involucradas en el proceso de biosíntesis de los esteroides corticosuprarrenales, puede faltar o tener constitución anómala, dando lugar a trastornos de carácter autosómico recesivos denominados hiperplasia suprarrenal congénita (HSC) y que puede producir modificaciones en la diferenciación sexual en ambos sexos, razón por la cual fue conocida con el nombre de síndrome adrenogenital (3). La forma más frecuente de hiperplasia suprarrenal congénita es aquella producida por deficiencia de la enzima 21-hidroxilasa, la cual abarca alrededor del 95% de todas las causas de esta entidad, correspondiendo 50% a la forma perdedora de sal y 50% a la forma simple virilizante (4,5). En nuestra casuística hemos encontrado que 55% pertenece a la forma simple virilizante y 45% a la forma perdedora de sal (6,7). La hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa presenta dos formas, una denominada clásica que puede ser perdedora de sal o virilizante simple y la forma no clásica. La forma perdedora de sal cursa con niveles bajos de cortisol y aldosterona, con pérdida de electrolitos y agua con deshidratación grave y colapso cardiocirculatorio, se observa los primeros días de vida post-natal; en la forma simple virilizante, las niñas presentan clitoromegalia, fusión en diverso grado de los labios y, en los casos severos, una abertura común para uretra y vagina, los varones presentan macrogenitosomia. La forma no clásica se presenta en la etapa peripuberal. El diagnóstico de HSC por deficiencia de 21- hidroxilasa se hace por cuantificación de la 17-hidroxi progesterona (17-OH-P) y cuando se sospecha de la forma perdedora de sal es importante la determinación del sodio y potasio sérico, los cuales se encuentran alterados aún antes de presentarse el cuadro clínico de deshidratación (7). En un estudio preliminar se ha encontrado que la hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa, observada en pacientes peruanos, el 50% de ellos presentaba la forma convencional de mutaciones y 50% parecerían corresponder a nuevas mutaciones diferentes a las investigadas por las técnicas empleadas (13). El propósito del presente trabajo es el dar a conocer el patrón genético de los pacientes peruanos y sus familiares portadores de hiperplasia suprarrenal congénita en sus variedades virilizante y perdedora de sal. Método Pacientes y familiares. Se han seleccionado 20 pacientes que fueron diagnosticados y seguidos en el Servicio de Endocrinología del Instituto de Salud del Niño, 13 padres, 13 madres y 5 hermanos para el estudio genético, cuando el padre estaba ausente se estudió un familiar de la línea paterna de sexo masculino abuelo o tío, en nuestro estudio se hizo con un abuelo paterno. Todos los pacientes fueron evaluados clínicamente, se les realizó determinaciones de 17-OH progesterona (17-OH-P), dehidroepiandrosterona-sulfato (DHA-S) y/o Δ4- androstenodiona y aquellos con la forma perdedora de sal se les dosó electrolitos (Na+ y K+) y cuando fue posible la actividad renina plasmática (PRA). A cada uno de ellos se le tomó una muestra de sangre en papel de filtro y las muestras de sangre seca enviadas al Laboratorio del Instituto de Salud Pública de la Ciudad de Sapporo (Japón) para el estudio genético mediante la técnica de PCR. Método de estudio genético. La técnica utilizada para el estudio genético fue la de la polymerase chain reaction (PCR) y se analizaron las mutaciones y delecciones más frecuentes encontradas tanto en Japón como en EE.UU. de N.A., como ya ha sido reportado previamente (13). Resultados De los 20 pacientes estudiados, 11 fueron de sexo femenino y 9 de sexo masculino; de otro lado, 16 pacientes eran portadores de la variedad perdedora de sal y 4 de la forma virilizante. Tres hermanos de los pacientes no estuvieron afectados y 2 presentaron alteraciones genéticas y cuadro clínico (nacieron cuando se realizaba este estudio). En la Tabla 1 se muestran los resultados de dosar las hormonas y electrolitos en pacientes con hiperplasia suprarrenal congénita de las variedades perdedores de sal y virilizante simple.
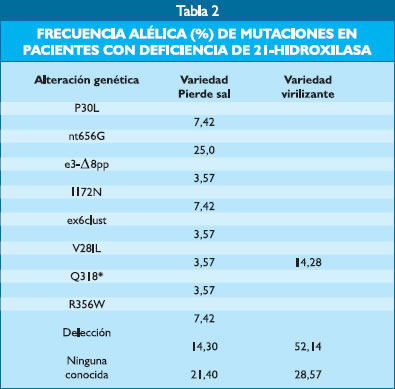
En la Tabla 2 se muestran los resultados de las principales mutaciones y deleciones observadas en 20 pacientes peruanos, sus padres y sus hermanos, de acuerdo a la forma clínica.
Discusión La hiperplasia suprarrenal congénita es una patología poco frecuente en endocrinología pediátrica. Ella se debe a defecto enzimático de carácter autosómico recesivo, que es potencialmente grave. Cada una de las enzimas que interviene en la génesis esteroidea puede producir hiperplasia suprarrenal congénita, siendo la más frecuente la deficiencia de 21-hidroxilasa, dando lugar a cuadros clínicos de diversa severidad y que pueden producir ambigüedad sexual (3,8,9,14,15,16). Se ha señalado que la incidencia de hiperplasia suprarrenal congénita debida a deficiencia de 21-hidroxilasa es de 1:10 000 a 1: 17 000 en Europa occidental y en los Estados Unidos de N.A. y a escala mundial sería de 1: 14 000 recién nacidos vivos (17), aún cuando otros señalan una prevalencia de 1:5 000 a 1:15 000 recién nacidos vivos (18). En la actualidad se dispone de métodos de tamizaje neonatal de hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa, que permite la detección precoz de esta entidad y realizar asignación del sexo en forma correcta y temprana, así como iniciar tratamiento precoz de los casos perdedores de sal. En Suecia se ha podido determinar mediante tamizaje neonatal una prevalencia de 1: 9800 (19), en Nueva Zelanda el tamizaje neonatal de deficiencia de 21-hidroxilasa arroja una frecuencia de 1 caso en 23,344 (20). En el Perú el tamizaje para detección precoz de recién nacidos con hiperplasia suprarrenal congénita se inició en el Instituto Especializado Materno Perinatal, quienes señalan que entre octubre del 2003 a agosto del 2005 se tamizaron 26451 recién nacidos obteniéndose dos casos positivos, lo que correspondería a una incidencia para hiperplasia suprarrenal congénita de 1/13225 recién nacidos vivos (21). Algunas veces se hace necesario realizar la corrección de los niveles de 17-0H-Progesterona en relación a los pesos de nacimiento a fin de disminuir los falsos positivos (22). En el Instituto de Salud del Niño se ha determinado que existe una frecuencia de 3 casos por cada 1000 nuevas consultas (7). Se ha señalado que el gen que gobierna a la enzima 21-hidroxilasa se encuentra en el brazo corto del cromosoma 6 y está vinculado a los antígenos de histocompatibilidad (HLA), así como al complemento 4 y la properdina (8). Existen dos tipos de genes para la 21-hidroxilasa, uno activo, el CYP21 y otro inactivo o putativo, el CYP21P, los cuales se encuentran situados el uno al costado del otro, en tandem, con el factor de complemento 4: C4/CYP21 y C4/CYP21P. En la HSC por deficiencia de 21-hidroxilasa ocurre desequilibrio de ligamiento entre los genes que gobiernan dicha enzima y ciertos tipos de HLA (9), habiéndose encontrado, además, la existencia de micro o macroconversiones o delecciones del gen CYP21P, convirtiéndose el gen CYP21, activo, en una forma inactiva (10). Mediante técnica de PCR (polymerase chain reaction) se pueden determinar las principales mutaciones y delecciones, que dan origen a esta entidad, que ocurren a nivel del gen de 21-hidroxilasa (11,12). La afección se produce por una alteración de la enzima CYP21OH, la cual impide la progresión en la biogénesis del cortisol, en el caso de las formas virilizantes simples, y del cortisol y la aldosterona en el caso de las formas perdedoras de sal; el exceso de andrógenos suprarrenales es responsable de las manifestaciones de virilización que presentan las niñas. Dupont et al. (23) fueron los primeros en señalar que la deficiencia de 21-hidroxilasa tenía una estrecha relación genética con los HLA, debido a la alta coincidencia de alguno de ellos con esta entidad. Estudios posteriores demostraron que tanto el gen para la 21-hidroxilasa como el de los antígenos de histocompatibilidad se encontraban en el brazo corto del cromosoma 6, lo que explica su relación patogénica. Así mismo, se determinó la existencia de dos genes para el CYP21OH, uno inactivo, el CYP21P y otro activo el CYP21, estrechamente vinculados al factor del complemento C4 y a la properdina. La hiperplasia suprarrenal congénita se produce al ocurrir recombinaciones frecuentes, que producen duplicaciones, delecciones, conversiones genéticas, o intercambios entre los genomas C4A/CYP21P y C4B/CYP21, los cuales hacen que el gen CYP21 se torne no funcional. Debe destacarse que a cada forma clínica de hiperplasia suprarrenal congénita corresponde una alteración genética (24); dichas alteraciones genéticas son escasas y existe correlación entre la alteración encontrada y el grado de compromiso de la actividad 21-hidroxilasa en niños diagnosticados mediante tamizaje neonatal (25). Se han descrito diversas mutaciones que afectan el gen CYP21OH y que dan lugar a las diferentes formas de HSC por déficit de 21-hidroxilasa. Nuestros resultados muestran que los pacientes portadores de HSC, variedad perdedora de sal, presentan como alelo más frecuente al nt656G (25%), en menor grado los alelos P30L (7,42%), I172N (7,42%) y R356W (7,42%), los cuales también son frecuentes en la población japonesa; se encontró 14,30% de delecciones y 21,40% que no correspondían a las mutaciones mas frecuentes encontrados en Estados Unidos y el Japón. En la variedad virilizante simple se encontró que el alelo V281L fue de 14,28%, 52,14% correspondió a delecciones y 28,57% a ninguna conocida. Las mutaciones no encontradas en los pacientes estudiados pensamos se trate de delecciones o conversiones genéticas mayores, para lo que se requiere estudios genéticos con primers específicos. Es interesante remarcar que el patrón genético encontrado en nuestros pacientes y sus familiares próximos es similar a lo encontrado en pacientes japoneses, lo cual abogaría a favor del origen asiático del hombre peruano. Referencias Bibliográficas
1Profesor Emérito de la Facultad de Medicina Hipólito Unanue de la Universidad Nacional Federico Villareal, Lima - Perú. 2Servicio de Endocrinología del Instituto Nacional de Salud del Niño, Lima - Perú. 3Profesor Principal Facultad de Medicina Hipólito Unanue de la Universidad Nacional Federico Villareal, Lima - Perú. 4Servicio de Neonatología del Hospital E. Rebagliati Martens (IPSS), Lima - Perú. 5Instituto de Salud Pública de la Ciudada de Sapporo, Hokkaido, Sapporo, Japón. |
|||