|
|
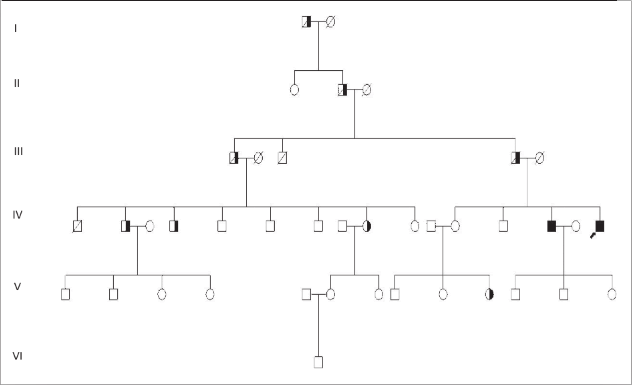
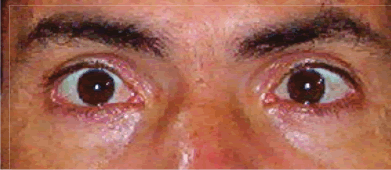
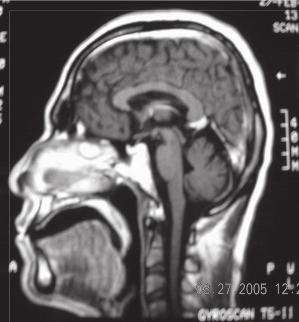
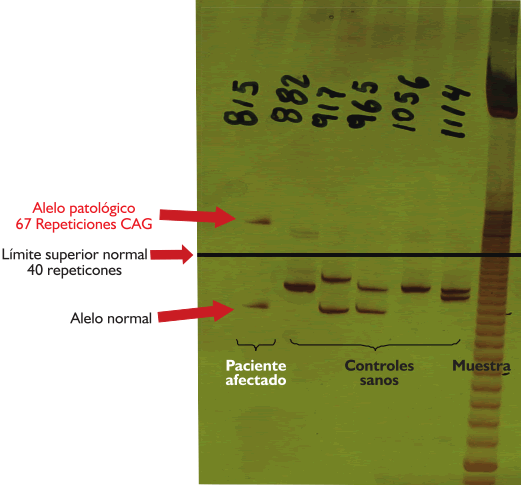
| Ataxia espinocerebelosa tipo 3 (Enfermedad de Machado Joseph) A propósito de un caso Luis Torres Ramírez(1),Miriam Vélez Rojas (1), Pilar Mazzetti Soler(2), Rafael Suárez Reyes (1), Carlos Cosentino Esquerre(1),Nicanor Mori Quispe(3), Mario Cornejo Olivas (2), María Marca Isabel(2), Olimpo Ortega Dávila(2), Miriam Villanueva Baltuano (4) Resumen Introducción: La enfermedad de Machado Joseph (MJD), o ataxia espinocerebelosa tipo 3 es un trastorno hereditario autosómico dominante ligado al gen de la ataxina 3 y se manifiesta en forma de ataxia progresiva, piramidalismo, oftalmoplejia, neuropatía, distonía y síndrome de piernas inquietas. Caso clínico: Varón de 34 años con ataxia progresiva, oftalmoplejia, y neuropatía periférica. Presenta antecedentes familiares con patrón autosómico dominante de trastorno similar en padre, abuelo paterno, tío, primos, sobrina y hermano. El estudio genético evidenció un alelo expandido de 67 repeticiones en heterocigosis en el gen de la SCA3. Conclusión: Presentamos el primer caso en Perú de un paciente con las características hereditarias, clínicas, neurofisiológicas y moleculares correspondientes a la enfermedad de Machado Joseph. Palabras clave: Ataxia, Enfermedad de Machado Joseph, SCA3. Abstract Introduction: Spinocerebellar ataxia type 3 (Machado Joseph disease) is an inherited autosomal dominant disorder related to the ataxina gene. Progressive ataxia, pyramidal sígns, ophthalmoplegia, peripheral neuropathy, dystonia and restless legs syndrome are main clinical features. Case report: A 34 year-old man with progressive ataxia, ophthalmoplegia and peripheral neuropathy. Positive familial antecedents with autosomal dominant pattern and included the father, grandfather, uncle, cousins and a sibling. Genetic study showed an expanded allele with 67 repeats in the SCA3 gene. Conclusion: We present the first peruvian patient with clinical, inheritance, neurophisiology and molecular features of Machado Joseph Disease. Key words: Ataxia, Machado Joseph disease, SCA 3. Introducción La enfermedad de Machado Joseph (MJD) o Ataxia Espinocerebelosa tipo 3 (SCA3), es un trastorno autosómico dominante, causado por la expansión inestable del triplete CAG del gen de la ataxina 3 ubicado en el cromosoma 14q24.3-q32.2. Las estructuras comprometidas a nivel del sistema nervioso central son los núcleos dentados y el pedúnculo cerebeloso superior, pudiendo afectarse los núcleos pontinos, los cordones posteriores, los haces espinocerebelosos y el núcleo dorsal de Clarke. El compromiso del sistema nervioso periférico (SNP) puede manifestarse como una neuropatía axonal. Clínicamente existen diversos fenotipos relacionados al número de repeticiones, es la ataxia cerebelosa el signo característico. Presentamos el caso de un paciente con las manifestaciones hereditarias, clínicas, neurofisiológicas y moleculares correspondientes a la enfermedad de Machado Joseph. Caso clinico Varón de 45 años, natural y procedente de Piura, que desde los 20 años presenta cuadro progresivo de ataxia para la marcha, diplopía horizontal binocular, dolor osteomuscular difuso, disminución de fuerza a predominio distal y torpeza motora, disfagia e hipofonía. Su padre, abuelo paterno, tío, primos, sobrina y hermano presentaron enfermedad similar (Figura 1). En la exploración neurológica se evidenció: funciones corticales normales; fuerza muscular conservada; tono muscular sin alteraciones; atrofia muscular global y simétrica; hiperreflexia generalizada; Babinski derecho; sensibilidad superficial y profunda conservada; fundoscopia normal; compromiso de VI nervio craneal izquierdo en posición primaria de la mirada (Figura 2) y de la mirada conjugada horizontal; no dismetría, discronometría o adiadococinesia aunque con marcha atáxica. La Resonancia Magnética (RM) de encéfalo informó atrofia bulbar y del vermis cerebeloso con aumento del cuarto ventrículo (Figura 3). Los estudios neurofisiológicos mostraron una polineuropatía sensitiva axonal en cuatro extremidades, presencia de unidades motoras polifásicas con características neurogénicas crónicas y potenciales de acción muscular normales. La evolución clínica fue estacionaria, lográndose alivio de dolor osteomuscular con tratamiento sintomático y terapia física. En el estudio genético para SCA3 se evidenció un alelo expandido de 67 repeticiones en heterocigosis en el gen de la SCA3 en nuestro paciente y en el hermano (Figura 4).
Discusión Descrita por primera vez en 1972 en una familia de inmigrantes portugueses en Massachussets (1), MJD pertenece al grupo de enfermedades de la poliglutamina caracterizadas por expansión inestable de trinucleótidos CAG, debido a una alteración en el gen de la ataxina 3 ubicado en el cromosoma 14q24.3-q32.2 (2,3). Es el subtipo más común entre las SCA, seguido por la SCA2 y la SCA6 (3). Su fenotipo es el más variable, manifestándose en forma de ataxia cerebelosa para la marcha y postura, alteraciones oculomotoras, signos piramidales, disartria, distonía, parkinsonismo, espasticidad, neuropatía, y síndrome de piernas inquietas (4,5). El caso presentado manifestó como síntoma inicial ataxia cerebelosa progresiva para la marcha. Anatomopatológicamente, la ataxia para la marcha en pacientes con SCA3 se debería al compromiso de los circuitos cerebelo-tálamo-cortical y tálamo-cortical motor (2), así como del núcleo reticular lateral del bulbo, núcleo precerebeloso encargado de la propiocepción y de automatismos somatomotores (6). Son frecuentes los disturbios del sueño, como el síndrome de piernas inquietas y los movimientos periódicos de las piernas (7). También son frecuentes la intolerancia al frío y la nicturia. El pseudoexoftalmo, las mioquimias faciolinguales y la distonía han sido descritos pero no son específicos (8). El paciente tenía retracción de párpados, característica del pseudoexoftalmo y alteración de motilidad ocular. El compromiso de los movimientos oculares sacádicos en los pacientes con SCA3 estaría en relación con la degeneración no sólo de los núcleos de nervios craneales sino también de un probable compromiso del núcleo interpuesto del rafe, núcleo rostral intersticial del fascículo longitudinal medial y núcleo retículo-tegmental protuberancial (9). En MJD los estudios por RM de encéfalo permiten observar la atrofia infratentorial, principalmente una degeneración selectiva de la protuberancia cerebral con compromiso temprano del tegmentum, mientras que la base permanece en el rango normal mucho tiempo después del inicio de los síntomas (10). En este paciente se evidenciaron atrofia bulbar, protuberancial y cerebelosa vermiana así como agrandamiento del cuarto ventrículo. Los estudios neurofisiológicos del 75% de los pacientes con MJD demuestran una neuropatía axonal sensitiva y motora y el enlentecimiento de la velocidad de conducción nerviosa (VCN), a consecuencia del compromiso inicial del SNC (8), especialmente en pacientes con menos de 73 repeticiones del triplete CAG, quienes mayormente desarrollan neuropatía (11). En este paciente se encontró una polineuropatía axonal sensitiva, mientras que la VCN motora fue normal. La presencia de unidades motoras polifásicas con características neurogénicas crónicas y con potenciales de acción muscular normales sugieren un compromiso difuso de células del asta anterior. En Latinoamérica Orosco Díaz y col. (12) estudiaron varias familias afectadas en Holguín, Cuba, y Gispert y col. (13) realizaron posteriormente el estudio genético, calificando a las familias afectadas como casos de ataxia espinocerebelosa tipo 2 (SCA-2), identificando el defecto en el locus genético 12q23.24. En Perú se publicó un estudio clínico y genético molecular en una familia afectada por ataxia espinocerebelosa tipo 7 (SCA-7), ligada a la mutación del brazo corto del cromosoma 3 (14). Este es el primer caso reportado en el Perú de un paciente con las características hereditarias, clínicas, neuro- fisiológicas y moleculares correspondientes a la enfermedad de Machado Joseph. Referencias Bibliográficas
1 Unidad de Movimientos Involuntarios, Instituto Nacional de Ciencias Neurológicas. 2 Unidad de Neurogenética. Instituto Nacional de Ciencias Neurológicas. 3 Médico Neurólogo de asistencia libre. 4 Médico de asistencia libre. |
||||||||