|
|
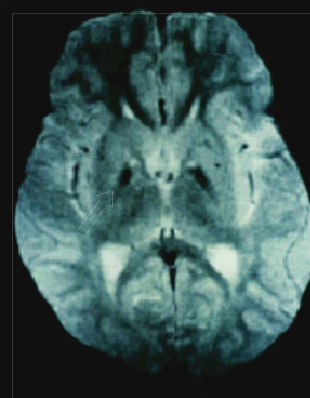
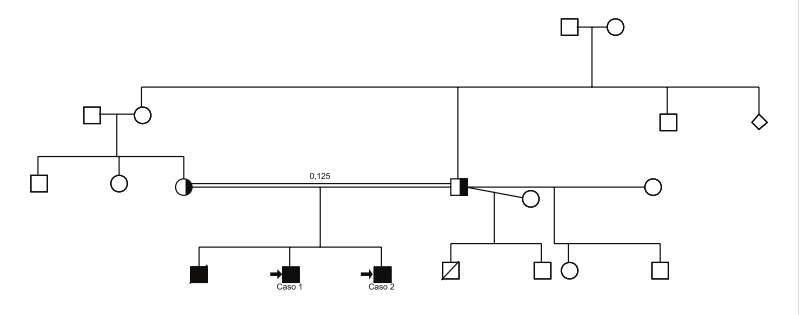
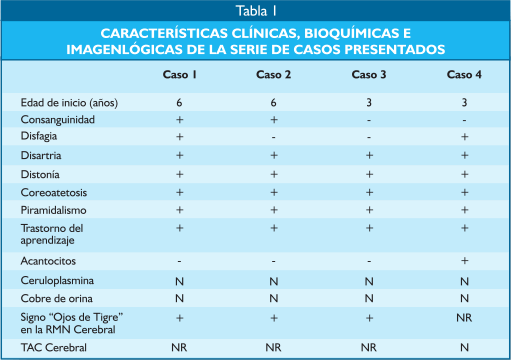
| Probable neurodegeneración asociada a pantotenato kinasa (PKAN)Estudio de cuatro casos en Perú Luis Torres Ramírez(1), Carlos Cosentino Esquerre (1), Nicanor Mori Quispe (2), , Miriam Vélez Rojas (1) ResumenIntroducción: La PKAN (Neurodegeneración Asociada a Pantotenato Kinasa) es una enfermedad autosómica recesiva caracterizada clinicamente por presentarse en la primera infancia, regresión en el desarrollo psicomotor, compromiso extrapiramidal, piramidal y neuro-oftalmológico, de curso progresivo y fatal. La alteración genética se encuentra en el cromosoma 20p12.3-p13 que codifica la pantotenato kinasa, que regula el metabolismo del pantotenato, fundamental en la síntesis de ácidos grasos. Su alteración resulta en un aumento de la concentración de cisteína y el depósito de hierro en los ganglios basales. Casos Clínicos: Se presentan cuatro pacientes, dos de ellos con antecedente de consanguinidad, que iniciaron síntomas neurológicos entre los 3 a 6 años de edad, caracterizados por distonía, coreoatetosis, regresión psicomotora, compromiso visual y piramidalismo, de curso progresivo, que fallecieron antes de los 10 años de edad. La RM de encéfalo mostró las características imágenes de “ojos de tigre”, correspondientes al depósito de hierro en los globos pallidus, concluyéndose en el diagnóstico de PKAN. Conclusión: Se presentan cuatro pacientes peruanos que por sus características hereditarias, clínicas y radiológicas corresponden al diagnóstico de PKAN. Palabras Clave: Herencia autosómica recesiva, PKAN, regresión psicomotora, signo de “ojos de tigre”. AbstractIntroduction: PKAN is an autosomal recessive disorder clinically characterized by delayed motor involvement, pyramidal and extrapyramidal symptoms, and neuro-ophthalmologic involvement with a progressive course and fatal outcome. The causal gene is localized to chromosome 20p12.3-p13 which encodes for panthothenate kinase. Pantothenate kinase regulates panthothenate metabolism and is integral to fatty acids synthesis. Its alteration may result in an elevated concentration of cysteine and iron accumulation in the basal ganglia. Case Report: We report four patients between the ages of 3 and 6 with a history of consanguinity who presented with a specific constellation of neurological symptoms, characterized by dystonia, choreoatetosis, psychomotor regression, visual involvement, pyramidalism, and a progressive course culminating in death prior to the age of 10. MRI showed the “eye-of-the-tiger” sign related to iron deposits at the globus pallidus, consistent with the diagnosis of PKAN. Conclusion: We report four Peruvian cases that according to inheritance, clinical and imaging characteristics correspond with a diagnosis of PKAN. Keywords: Autosomic recessive, extrapyramidal and pyramidal signs, “eye-of-the-tiger” sign, PKAN, psychomotor regression . IntroducciónLa Neurodegeneración Asociada a Pantotenato Kinasa (PKAN por sus siglas en inglés), anteriormente llamada Enfermedad de Hallervorden - Spatz (EHS) (1-3), es un desorden autosómico recesivo de inicio en la infancia temprana, entre los 3 y 6 años de edad, caracterizado por regresión del desarrollo psicomotor, distonía, coreoatetosis, disartria, espasticidad, retinopatía bilateral, atrofia óptica y convulsiones, de curso progresivo (4). El gen causal fue localizado en 1996 por Taylor et al. en el cromosoma 20p12.3-p13 (5). En el 2001, Zhou (6) aísla la región crítica e identifica el gen PANK2 que codifica la pantotenato kinasa. En la anatomía patológica hay atrofia cerebral, con depósitos de pigmentos de hierro alrededor de los vasos sanguíneos en el globus pallidus y substancia nigra pars reticulata, numerosos axones esferoidales, observados en todo el SNC, especialmente en los ganglios basales y el tronco cerebral, así como también ovillos neurofibrilares y cuerpos de Lewy (7). Los pigmentos que contienen hierro se acumulan en gran cantidad en estas áreas, y pueden contribuir a la distrofia axonal (8). La enfermedad ha sido observada en diversas partes del mundo (9-11). En América Latina, se han reportado casos en México (12,13), Brasil (14,15) y Venezuela (16). Presentamos cuatro pacientes, que desde el punto de vista hereditario, clínico e imagenológico corresponden a la PKAN, siendo los primeros casos comunicados en Perú. Casos clínicosCaso 1: Paciente Ed... VS, de 8 años de edad, con antecedente de consanguinidad paterna, tuvo dos hermanos con enfermedad similar (Figura 1), uno de ellos fallecido. Desarrollo psicomotor normal hasta los 6 años de edad en que presentó movimientos involuntarios de tipo contracciones sostenidas en cuello y en miembro superior derecho a predominio distal, meses después se extiende al miembro superior izquierdo y miembros inferiores, comprometiendo la marcha. Además, movimientos de rotación del cuello y movimientos periorales que dificultan la articulación de la palabra. A los ocho años camina con apoyo, seis meses más tarde queda postrado. Recibió dosis altas de tetrazepam y trihexifenidil con leve mejoría. En el examen neurológico presentó distonía severa del cuello y extremidades, con movimientos coreoatetósicos, hiperreflexia osteotendinosa generalizada. El electroencefalograma (EEG) evidenció actividad delta difusa, irregular, no paroxismal. La evaluación neuropsicológica informó retardo mental profundo. En la tomografía axial computada (TC) cerebral se observó hiperdensidad en los ganglios basales en forma bilateral; la resonancia magnética nuclear (RMN) cerebral mostró los “ojos de tigre” (Foto 1). La radiografía de tórax, serología para lúes, estudio de HIV fueron negativos. La ceruloplasmina y el dosaje de cobre en orina fueron normales y no se encontró anillo de Kayser-Fleisher en el estudio con lámpara de hendidura.
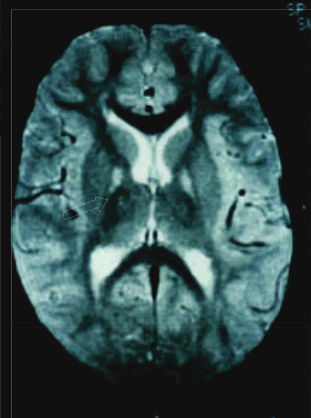
Caso 2: Paciente Es... VS de 7 años de edad, hermano del paciente del Caso 1, inicia síntomas a los 6 años, con dificultad para deambular por presentar contracciones sostenidas en miembros inferiores, poco tiempo después afecta los miembros superiores, de curso progresivo hasta la postración, dificultad para deglutir y articular palabras. En el examen neurológico presentó disartria e imposibilidad para la marcha, con posturas distónicas en miembros superiores y cara, movimientos coreoatetósicos en manos; hiperreflexia osteotendinosa, signo de Babinski bilateral. La RMN mostró marcada hiperseñal de ambos globos pálidos en secuencia T1 e hiposeñal en T2 (Foto 2).
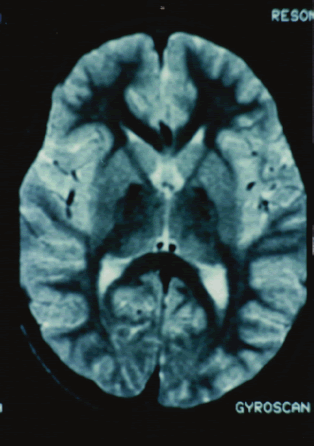
Caso 3: Paciente MAD, de sexo femenino, de 7 años, sin antecedente de consanguinidad ni familiares con enfermedad similar. La historia de la gestación y perinatal fue normal. A los tres años de edad, cuando ya deambulaba, presenta contracciones sostenidas en miembros inferiores y superiores que la obliga a adoptar posturas anormales que dificultan sus actividades de vida diaria y condicionan caídas continuas. Poco tiempo después dificultad en la articulación de la palabra. Desde los tres años tendencia a mantener el tronco en hiperextensión y los miembros superiores en rotación externa con flexión de los dedos que se acentúan al realizar un movimiento voluntario; dificultad para la visión en la oscuridad. Recibió tetrazepam y trihexifenidil por progresión del movimiento anormal. Al examen neurológico, paciente alerta, colaboradora, con disartria, movimientos distónicos y coreoatetosicos en las cuatro extremidades, tronco y en región perioral; hiperreflexia generalizada con clonus plantar, signo de Babinski bilateral presente, sin atrofia muscular. Las evaluaciones de neuro-oftalmología, ceruloplasmina, EEG y bioquímica sanguínea fueron normales. Se encontró un 25% de acantocitos en sangre periférica. La RMN en secuencia T2 evidenciaba marcada hiposeñal de ambos globos pálidos y menor acentuación en sustancia negra (Foto 3).
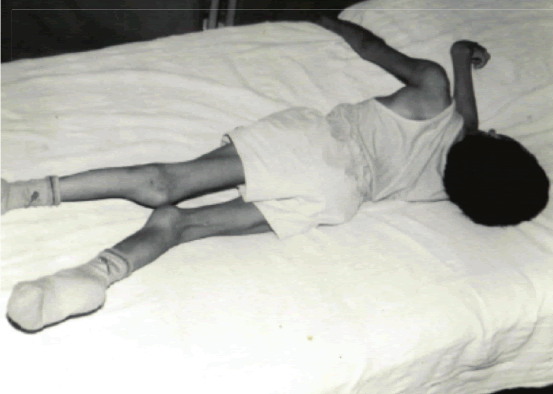
Caso 4: Paciente PCV, de 7 años de edad, sin antecedentes de consanguinidad, tiene dos hermanos sanos y uno menor que sufre de convulsiones. Desde los tres años de edad presenta retardo psicomotor y del lenguaje. La familia refería: “no tenía estabilidad, se caía con frecuencia y era muy llorón”, irritable, inquieto, inicia su marcha a los cuatro años pero observan que hemicuerpo izquierdo tiene “menos fuerza” y tiene dificultad para articular palabras. Dos meses antes de hospitalización notan movimientos involuntarios en miembros superiores a predominio izquierdo y que adopta actitudes especiales para compensar el movimiento. Informan que el niño tiene dificultad para la visión nocturna. Al examen neurológico presentó disartria, comprende órdenes simples, hiperquinético, con dificultad para mantenerse de pie, amplía la base de sustentación, permanece con los miembros superiores fijados al tronco por la presencia de movimientos coreoatetósicos, y actitud distónica del tronco y cuello (Foto 4). En el fondo de ojo se encuentra hipoplasia neuroretinal. La evaluación neuropsicológica encuentra retardo mental leve con un CI 66. En el EEG se observan espigas aisladas de localización bitemporooccipital. La TC cerebral fue normal.
Hallervorden y Spatz en 1922 estudiaron una familia de doce miembros, donde cinco hermanas presentaron entre los 7 a 9 años, dificultad para la marcha, rigidez, distonía, deforma-ción de miembros inferiores, disartria, disfagia, movimientos coreatetósicos, deterioro intelectual progresivo y compromiso del tracto corticoespinal; que fallecieron entre los 16 y 27 años (1). Dooling en 1974 (17), estableció las características clínicas y la anatomía patológica del Síndrome de Hallervorden Spatz (SHS) en su estudio de 64 individuos: Inicio en la niñez, desorden motor extrapiramidal con posturas distónicas, rigidez, movimientos involuntarios, compromiso de la vía corticoespinal, deterioro cognitivo, curso progresivo en años y fallecimiento al inicio de la edad adulta. El origen de esta enfermedad es genético con el locus identificado a nivel del cromosoma 20p12.3–p13 (5), y cuyo gen alterado es el PANK2 el cual codifica la pantotenato kinasa (6). Las mutaciones en este gen se han encontrado en pacientes con el síndrome típico y en aquellos con fenotipos variables. En nuestros pacientes, el diagnóstico genético no se realizó. La pantotenato kinasa es la enzima reguladora clave en la síntesis de la coenzima A proveniente del pantotenato, y es así que el defecto en el metabolismo del pantotenato es a su vez fundamental para la síntesis de ácidos grasos y el metabolismo energético. El gen PANK2 se expresa en la retina y en los ganglios basales y al haber una disminución de la producción de coenzima A en estos tejidos resultará en una biosíntesis defectuosa de las membranas celulares (4). La producción reducida del 4-fosfopantotenato puede resultar en un incremento de la concentración de la cisteína, la cual condensa al fosfopantotenato en los ganglios basales, y ser ésta la responsable de la acumulación de hierro en estas áreas con el complejo hierro-cisteína ocasionando daño de las membranas neuronasles y el ADN mediante generación de radicales libres y la promoción del estrés oxidativo (18). El compromiso familiar está presente en aproximadamente la mitad de los pacientes, y el 15% de los pacientes tienen alguna relación de consanguinidad o son casos esporádicos (13). Los casos 1 y 2 eran hermanos, sus padres tenían relación de consanguinidad y tuvieron un hermano mayor afectado fallecido (Figura 1). Los casos 3 y 4 son de presentación esporádica y no hubo relación de consaguinidad.
Las manifestaciones del SNC varían de paciente en paciente. El cuadro clínico incluye rigidez progresiva, anormalidades posturales, distonía, disartria, coreoatetosis, temblor. La hiperreflexia, espasticidad y respuesta extensora plantar son frecuentes. El deterioro intelectual es progresivo (19). La atrofia óptica puede ser el primer y por un tiempo, el único síntoma de la EHS (20). Retinitis pigmentaria y convulsiones suelen también estar presentes (21, 22). La forma clásica se presenta al término de la primera década; el compromiso corticoespinal y la disfunción extrapiramidal, pueden desarrollarse durante varios años (23). Los cuatro pacientes presentaron compromiso corticoespinal y extrapiramidal, con regresión del desarrollo psicomotor, de curso progresivo. Sólo el Caso 4 presentó compromiso visual. La distonía severa, rigidez, dificultad en masticar, deglutir y compromiso respiratorio están presentes uno o dos años después del inicio de los primeros síntomas. Sin embargo, el curso lento y progresivo puede continuar hasta la tercera y cuarta década de vida. La progresión a la muerte al inicio de la edad adulta está descrita. Nuestros Casos 1, 2 y 4 fallecieron a los 9 años, desconociéndose la evolución del Caso 3. El estudio convencional de laboratorio es irrelevante, pero la RMN cerebral es el examen que más ha contribuido a la comprensión de la PKAN, siendo particularmente útil en separarla de otras formas de Neurodegeneración con Acumulación de Hierro Cerebral (NBIA por sus siglas en inglés) (24). En la PKAN, una región central de hiperintensidad con hipointensidad circundante del globus pallidus en T2 es virtualmente patognomónica. La zona de hipertintensidad representa cambios patológicos que incluyen gliosis, desmielinización, pérdida neuronal e inflamación axonal, mientras que la zona hipointensa es causada por la pérdida de señal secundaria a la deposición de hierro (25). Hayflick (19) estudió 123 pacientes con diagnóstico de SHS, y reportó que todos los pacientes con clínica típica y atípica que tuvieron el gen PANK2 mostraron imágenes en RMN conocidas como “ojos de tigre”, como en tres de los casos presentados. Si bien estos cambios se suceden una vez que se cuenta con el cuadro clínico completo, se ha reportado hiperintensidad del globus pallidus en estadios iniciales (26), lo cual sugiere que podrían ser diferentes manifestaciones de la enfermedad durante su evolución, pudiéndose encontrar a inicios de la enfermedad estudios de RMN normales y en estadíos finales disminución de la hiperintensidad, la cual puede incluso llegar a desaparecer (27). Los hallazgos clínicos y los encontrados en la RMN de nuestros cuatro pacientes son altamente sugestivos de EHS. Uno de nuestros pacientes mostró estos cambios de tipo evolutivo en la RMN. No existe tratamiento específico para el SHS. Los agentes quelantes para remover el exceso de hierro como la desferrioxamine no disminuye los depósitos de hierro y no afecta el curso clínico (4). La distonía puede ser tratada con levodopa-carbidopa, bromocriptina, solas o en combinación, con leve a moderado beneficio (28). El baclofeno mejora la espasticidad pero la fisoterapia rara vez es beneficiosa (29). La actividad convulsiva requiere la administración de drogas antiepilépticas, como carbamazepina o fenitoína (30). Al progresar la enfermedad, la disartria y la deglución son cada vez mayores requiriendo incluso de gastrostomía. En conclusión, comunicamos estos cuatro casos de pacientes peruanos por lo excepcional de su presentación. De inicio en la infancia, curso progresivo, con distonía generalizada, trastornos cognitivos, signos piramidales y extrapiramidales; las imágenes de RMN mostraron los característicos “ojos de tigre”. Con todos estos signos siempre la primera posibilidad diagnóstica será PKAN. A medida que la prueba que permita la detección de la mutación PANK2 esté disponible en nuestro medio, podrá ser usada para confirmarla y ofrecer diagnóstico prenatal a las familias afectadas (31). Referencias Bibliográficas
1Neurólogos del Departamento de Enfermedades Neurodegenerativas. Instituto de Ciencias Neurológicas, Lima, Perú. 2 Médico Neurólogo de Asistencia Libre. |
||||||||||||