|
|
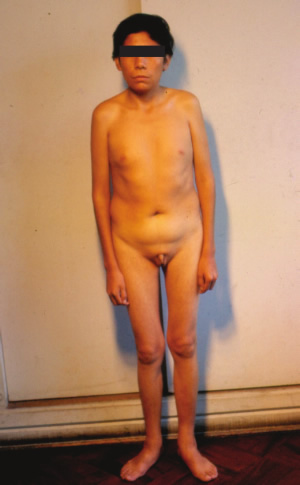
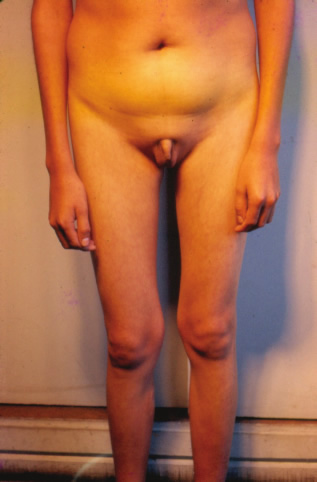
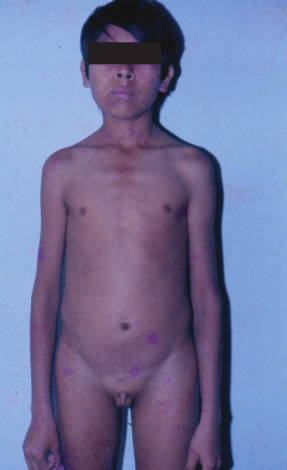
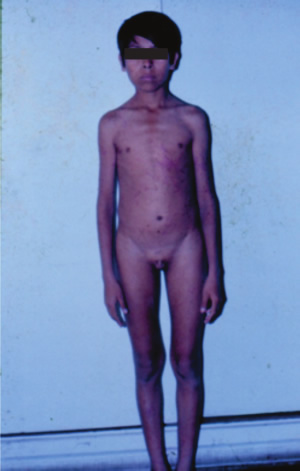
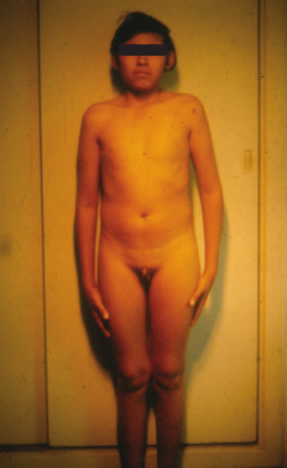
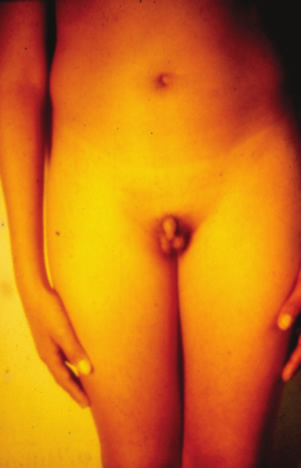
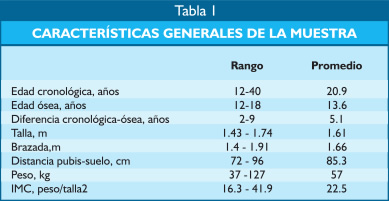
| Aportes al conocimiento del hipogonadismo hipogonadotrófico congénito Fausto Garmendia Lorena (1),Héctor Valdivia Carpio , Oscar Castillo Sayán (2,3) Resumen El hipogonadismo hipogonadotrófico congénito (HHC) es una forma clínica prepuberal de insuficiencia gonadal de origen hipotalámico por deficiencia congénita de la secreción de la hormona liberadora de gonadotrofinas (GnRH), vinculada a defectos cromosómicos. Material y métodos: Se describe 35 casos de HHC, 24 con alteración olfatoria correspondientes al síndrome de Kallmann, 21 varones y 3 mujeres y 11 con HH idiopático sin alteración olfatoria, 8 varones y 3 mujeres, de 12 a 40 años, promedio 20.9 años, que fueron evaluados clínicamente y mediante determinaciones de LH, FSH, testosterona (T), pruebas de GnRH e hipoglicemia insulínica, edad ósea, función tiroidea. Resultados: En todos se comprobó manifestaciones de hipogonadismo prepuberal, genitales infantiles, ausencia de caracteres sexuales secundarios, ausencia de interés y actividad sexual, amenorrea primaria en las 6 mujeres, proporciones eunucoides, edad ósea menor que la edad cronológica,concentraciones bajas de LH, FSH,T, las pruebas de GnRH e hipoglicemia fueron positivas, función tiroidea normal y cromatina sexual normal. Conclusiones: Se concluye que se trata de pacientes con hipogonadismo hipogonadotrófico hipotalámico congénito por deficiencia de GnRH. Palabras clave: Hipogonadismo hipogonadotrófico prepuberal, deficiencia congénita de GnRH, defectos olfatorios. Abstract The congenital hypogonadotropic hypogonadism (CHH) is a prepuberal clinical form of gonadal hypofunction due to a congenital hypothalamic deficiency of the gonadotropin releasing hormone (GnRH) related to chromosomial defects. Material and methods: A total of 35 cases of CHH, 24 with olfaction defects corresponding to the Kallman´s syndrome, 21 male and 3 female and 11 to the idiopathic hypogonadothrophic hypogonadism without olfactory defects, 8 male and 3 female, 12 to 40 years old, mean 20.9 years are described. Clinical evaluations as well as determinations of LH, FSH, testosterone (T), GnRH and insulin hypoglycemic tests, radiological bone age, thyroid function tests, sexual chromatin determinations were conducted. Results: All of them had clinical manifestations of prepuberal hypogonadism characterized by infantile genitalia, absence of secondary sexual characteristics, no sexual interest and activity, primary amenorrhea in the 6 female patients, eunuchoidal body proportions, bone age lower than the chronological one. These patients had low basal LH, FSH, T concentrations in blood, positive GnRH and hypoglycemia tests, normal thyroid function and normal sex chromatin. Conclusions: It is concluded that these patients suffered from a congenital hypothalamic hypogonadothropic hypogonadism due to GnRH deficiency. Key words: Prepuberal hypogonadotrophic hypogonadism, congenital GnRH deficiency, olfactory defects. Introducción El hipogonadismo hipogonadotrófico congénito (HHC)) es una forma clínica de hipogonadismo, de origen hipotalámico por deficiencia congénita de la secreción pulsátil de GnRH, de presentación prepuberal, mas frecuente en varones que en mujeres, que se acompaña de anosmia o hiposmia, constituyendo el síndrome de Kallman (2) o sin alteración olfatoria considerado como hipogonadismo hipogonadotrófico idiopático. Estos pacientes tienen genitales infantiles, ausencia de caracteres sexuales secundarios, proporciones corporales eunucoides, edad ósea menor a la cronológica, pueden acompañarse de malformaciones congénitas como sordera, hipospadias, criptorquidia, labio leporino, vinculados a mutaciones cromosomiales sin tener alteraciones numéricas de los cromosomas sexuales; la deficiencia de GnRH suele ser aislada, por lo que aparte de la deficiencia de gonadotropinas ocasionalmente no tienen otras alteraciones de la función hipotálamo-hipofisaria. Material y métodos Se estudió a 35 pacientes con HHC, atendidos en el Servicio de Endocrinología del Hospital Dos de Mayo entre los años 1965 a 1985 que, al momento de la primera consulta tenían entre 12 a 40 años, promedio 20.9 años. Se elaboró la historia clínica en la que se consignó datos sobre el desarrollo sexual, talla, peso, IMC (índice de masa corporal), proporciones corporales, exploración de la función olfatoria. Se determinó LH (n=35), FSH (n=22), testosterona (n=20) por métodos radioinmunológicos en condiciones basales y, en parte de ellos (n=31), después de la estimulación con 100 ug de GnRH, administrada en bolo por vía intravenosa. Prueba de hipoglicemia con insulina, 0.1 UI/kg de peso en inyección i.v. en bolo (n=16) para conocer la reserva hipofisaria de somatotropina y de ACTH/cortisol. La función tiroidea fue examinada mediante determinaciones de PBI, T4 o captación de I-131 Determinación de la edad ósea (n=19) por el método de Pyle y Waterhouse, determinación de la cromatina sexual nuclear o de la fórmula cromosómica. Resultados Todos los pacientes presentaron manifestaciones de hipogonadismo prepuberal, esto es, genitales infantiles, ausencia de caracteres sexuales secundarios (Figuras 1 a 6), ausencia de interés y actividad sexual; 24 presentaron anosmia o hiposmia (síndrome de Kallman), 21 varones y 3 mujeres, y 11 sin alteración olfatoria (hipogonadismo hipogonadotrópico idiopático), 8 varones y 3 mujeres; las 6 mujeres tuvieron amenorrea primaria. La talla fue normal y en el seguimiento de estos pacientes se constató que continuaron creciendo más allá de la edad límite normal para ambos géneros. La brazada fue mayor que la talla y la distancia pubis-suelo mayor que la distancia vértex-pubis, constituyendo las proporciones eunucoides. La edad ósea fue menor a la edad cronológica entre 2.5 a 9 años, promedio 5.1 años. Excepto 1 paciente con obesidad mórbida, la mayoría tuvo peso e IMC normales (Tabla I). De los pacientes con síndrome de Kallmann, 2 presentaron sordera, uno de ellos oligofrenia con función tiroidea normal y 2 criptorquidia y labio leporino un paciente con HH con función olfatoria normal, los 5 fueron varones.
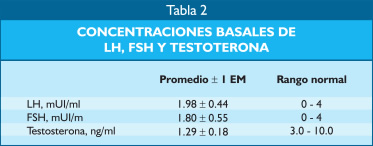
Las cifras basales de LH, FSH y testosterona se encontraron en rango bajo o normal bajo pese a tener cifras bajas de testosterona para la edad de los pacientes varones (Tabla 2).
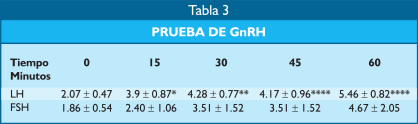
La administración de GnRH en bolo determinó una elevación de LH y FSH que, en la comparación con el valor basal, la diferencia fue estadísticamente significativa para LH desde los 15 a los 60 minutos, lo que demuestra que la alteración de la secreción de gonadotropinas era de origen hipotalámico (Tabla 3).
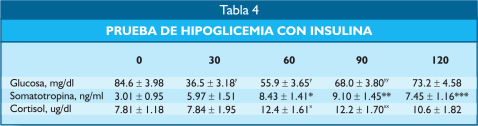
La administración de insulina, 0.1 UI/kg de peso, ocasionó una hipoglicemia suficiente como para elevar los valores de somatotropina y de cortisol en forma significativa, que demuestra que la función hipofisaria secretora tanto de hormona de crecimiento como de ACTH/cortisol estaban normales.
La función tiroidea fue normal en estos pacientes y el sexo cromosómico estuvo de acuerdo al sexo fenotípico y gonadal. Discusión La diversidad geográfica del Perú determina diferencias en el inicio de la pubertad, en las mujeres de nivel del mar la pubertad normalmente se inicia entre los 8 a 12 años y en las mujeres de altura entre los 10 a 14 años; los varones de nivel del mar, en cambio, inician la pubertad entre los 10 a 14 años y los de altura entre los 12 a 16 años (3-6). En los 35 pacientes estudiados, el desarrollo gonadal y de los caracteres sexuales secundarios no se presentaron en las edades señaladas anteriormente, constituyendo el síndrome de hipogonadismo prepuberal que, entre otras características, se acompañó de proporciones corporales eunucoides, la brazada mayor que la talla y el segmento pubis-suelo mayor que el segmento vertex-pubis, que se explica por la falta de producción de hormonas sexuales en la etapa puberal, que a su vez deriva de la deficiente producción de LH y FSH que es la consecuencia de la ausente secreción pulsátil de GnRH. Todo ello retarda la maduración ósea y el cierre de los cartílagos de crecimiento, fenómeno que guarda relación con el hallazgo del retardo en la edad ósea. Se ha podido documentar una diferencia entre edad cronológica y edad ósea mayor de 5 años y el hecho que las pacientes mujeres siguieron creciendo después de los 16 años y los varones después de los 18. A diferencia del hipogonadismo primario, que cursa con cifras elevadas de gonadotropinas, en el hipogonadismo hipogonadotrópico ellas están en concentraciones bajas o indetectables, pese a que las hormonas sexuales están en concentraciones bajas, tal como se encontró en los varones, cuyas cifras de testosterona estuvieron por debajo de 3 ng/ml: se encontró estradiol bajo en la única mujer en la que se midió esta hormona, pero todas ellas tuvieron amenorrea primaria. La elevación de las concentraciones de LH y FSH en respuesta a la administración de GnRH permite afirmar que la deficiencia hormonal de estos pacientes es de origen hipotalámico; se debe tener en cuenta, sin embargo, que en ocasiones es necesario efectuar más de una estimulación con GnRH para conseguir una respuesta apropiada de gonadotropinas. La elevación de las concentraciones de somatotropina durante la hipoglicemia insulínica y que los pacientes tuvieran una talla normal revelan que esta función hipofisaria estaba conservada; del mismo modo, la elevación de cortisol durante esta misma prueba demuestra que la función hipofiso-suprarrenal era normal. Todo ello, agregado a que la función tiroidea se encontraba normal permite concluir que la deficiencia de gonodotropinas, como está descrito para este tipo de condición patológica, es una deficiencia hipotalámica selectiva de la GnRH, que se sustenta, además, en la deficiencia del sentido de la olfacción, anosmia o hiposmia presentaron 24 de los 35 de los pacientes. La primera descripción de la concurrencia de hipogonadismo y anosmia se atribuye a Aureliano Maestre de San Juan, anatomista español, quien en1856 describió el caso de un varón adulto que en la autopsia tenía falta total de los nervios olfatorios con genitales infantiles y atrofia de los testículos (7,8); sin embargo este cuadro lleva el nombre del genetista y psiquiatra germano-norteamericano Franz Kallmann, quien en 1944 señaló el origen genético de esta patología (2). Se considera que el síndrome de Kallman se debe a un defecto en la etapa fetal tanto del desarrollo de los bulbos olfatorios como de la migración de las células productoras de GnRH a su ubicación normal en el hipotálamo. Estas alteraciones congénitas responden a 5 mutaciones genéticas hasta ahora conocidas con las denominaciones de KAL1, FGFR1, PROKR2, PROK2 y FGF8 (9), cuya diferente presencia y penetración explica la gran heterogeneidad de las manifestaciones clínicas de este síndrome(10,11), entre ellas la variante ahora conocida como HH idiopático, en la cual no existe alteración de la función olfatoria y que se debería a que las células productoras de GnRH en estos casos si migran normalmente del epitelio olfatorio al hipotálamo pero son incapaces para producir GnRH (8). Tal como está descrito, hemos hallado otras anomalías congénitas como criptorquidia, labio leporino, alteraciones variables de la audición, también pueden presentar hipospadias, paladar hendido, que cuando se presentan aparentemente solas es conveniente indagar sobre la posibilidad que formen parte del espectro clínico del hipogonadismo hipogonadotrópico congénito. Del mismo modo, se debe recordar que esta condición puede ser hereditaria y presentarse en varios miembros de una familia, inclusive con características diferentes, es decir, en una familia unos tener características de síndrome de Kallmann y otros de un HH idiopático, demostrando así que son variantes de un mismo desorden. Dentro de la complejidad de este cuadro clínico se ha descrito una forma de deficiencia de GnRH de inicio en la edad adulta, en la cual los pacientes presentan hipogonadismo con infertilidad pero que habían desarrollado sus caracteres sexuales normalmente en la pubertad (12). El tratamiento de esta condición patológica, que va mas allá del propósito de esta publicación, puede llevarse a cabo mediante la administración de hormonas sexuales testosterona y estrógenos en varones y mujeres respectivamente, o de gonadotropinas o de hormona coriónica gonadotrópica; pero la administración pulsátil de GnRH mediante una bomba de infusión es la mas apropiada desde que se puede conseguir no solo la normalización de la secreción de hormonas sexuales sino también de la capacidad reproductiva (13,14). Referencias Bibliográficas
1 Instituto de Investigaciones Clínicas. 2 Instituto Nacional de Biología Andina, Facultad de Medicina, Univesidad Nacional Mayor de San Marcos (UNMSM). 3 Hospital Nacional Arzobispo Loayza. |
||||||||||||||||